bamCoverage
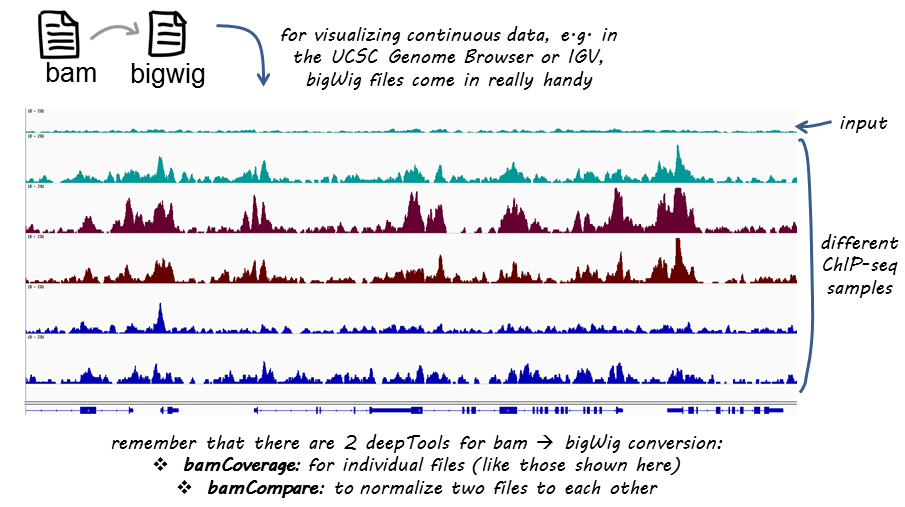
If you are not familiar with BAM, bedGraph and bigWig formats, you can read up on that in our Glossary of NGS terms
This tool takes an alignment of reads or fragments as input (BAM file) and generates a coverage track (bigWig or bedGraph) as output. The coverage is calculated as the number of reads per bin, where bins are short consecutive counting windows of a defined size. It is possible to extended the length of the reads to better reflect the actual fragment length. bamCoverage offers normalization by scaling factor, Reads Per Kilobase per Million mapped reads (RPKM), counts per million (CPM), bins per million mapped reads (BPM) and 1x depth (reads per genome coverage, RPGC).
usage: bamCoverage -b reads.bam -o coverage.bw
help: bamCoverage -h / bamCoverage --help
Required arguments
- --bam, -b
BAM file to process
Output
- --outFileName, -o
Output file name.
- --outFileFormat, -of
Possible choices: bigwig, bedgraph
Output file type. Either “bigwig” or “bedgraph”.
Optional arguments
- --scaleFactor
The computed scaling factor (or 1, if not applicable) will be multiplied by this. (Default: 1.0)
- --MNase
Determine nucleosome positions from MNase-seq data. Only 3 nucleotides at the center of each fragment are counted. The fragment ends are defined by the two mate reads. Only fragment lengthsbetween 130 - 200 bp are considered to avoid dinucleosomes or other artifacts. By default, any fragments smaller or larger than this are ignored. To over-ride this, use the –minFragmentLength and –maxFragmentLength options, which will default to 130 and 200 if not otherwise specified in the presence of –MNase. NOTE: Requires paired-end data. A bin size of 1 is recommended.
- --Offset
Uses this offset inside of each read as the signal. This is useful in cases like RiboSeq or GROseq, where the signal is 12, 15 or 0 bases past the start of the read. This can be paired with the –filterRNAstrand option. Note that negative values indicate offsets from the end of each read. A value of 1 indicates the first base of the alignment (taking alignment orientation into account). Likewise, a value of -1 is the last base of the alignment. An offset of 0 is not permitted. If two values are specified, then they will be used to specify a range of positions. Note that specifying something like –Offset 5 -1 will result in the 5th through last position being used, which is equivalent to trimming 4 bases from the 5-prime end of alignments. Note that if you specify –centerReads, the centering will be performed before the offset.
- --filterRNAstrand
Possible choices: forward, reverse
Selects RNA-seq reads (single-end or paired-end) originating from genes on the given strand. This option assumes a standard dUTP-based library preparation (that is, –filterRNAstrand=forward keeps minus-strand reads, which originally came from genes on the forward strand using a dUTP-based method). Consider using –samExcludeFlag instead for filtering by strand in other contexts.
- --version
show program’s version number and exit
- --binSize, -bs
Size of the bins, in bases, for the output of the bigwig/bedgraph file. (Default: 50)
- --region, -r
Region of the genome to limit the operation to - this is useful when testing parameters to reduce the computing time. The format is chr:start:end, for example –region chr10 or –region chr10:456700:891000.
- --blackListFileName, -bl
A BED or GTF file containing regions that should be excluded from all analyses. Currently this works by rejecting genomic chunks that happen to overlap an entry. Consequently, for BAM files, if a read partially overlaps a blacklisted region or a fragment spans over it, then the read/fragment might still be considered. Please note that you should adjust the effective genome size, if relevant.
- --numberOfProcessors, -p
Number of processors to use. Type “max/2” to use half the maximum number of processors or “max” to use all available processors. (Default: 1)
- --verbose, -v
Set to see processing messages.
Read coverage normalization options
- --effectiveGenomeSize
The effective genome size is the portion of the genome that is mappable. Large fractions of the genome are stretches of NNNN that should be discarded. Also, if repetitive regions were not included in the mapping of reads, the effective genome size needs to be adjusted accordingly. A table of values is available here: http://deeptools.readthedocs.io/en/latest/content/feature/effectiveGenomeSize.html .
- --normalizeUsing
Possible choices: RPKM, CPM, BPM, RPGC, None
Use one of the entered methods to normalize the number of reads per bin. By default, no normalization is performed. RPKM = Reads Per Kilobase per Million mapped reads; CPM = Counts Per Million mapped reads, same as CPM in RNA-seq; BPM = Bins Per Million mapped reads, same as TPM in RNA-seq; RPGC = reads per genomic content (1x normalization); Mapped reads are considered after blacklist filtering (if applied). RPKM (per bin) = number of reads per bin / (number of mapped reads (in millions) * bin length (kb)). CPM (per bin) = number of reads per bin / number of mapped reads (in millions). BPM (per bin) = number of reads per bin / sum of all reads per bin (in millions). RPGC (per bin) = number of reads per bin / scaling factor for 1x average coverage. None = the default and equivalent to not setting this option at all. This scaling factor, in turn, is determined from the sequencing depth: (total number of mapped reads * fragment length) / effective genome size. The scaling factor used is the inverse of the sequencing depth computed for the sample to match the 1x coverage. This option requires –effectiveGenomeSize. Each read is considered independently, if you want to only count one mate from a pair in paired-end data, then use the –samFlagInclude/–samFlagExclude options. (Default: None)
- --exactScaling
Instead of computing scaling factors based on a sampling of the reads, process all of the reads to determine the exact number that will be used in the output. This requires significantly more time to compute, but will produce more accurate scaling factors in cases where alignments that are being filtered are rare and lumped together. In other words, this is only needed when region-based sampling is expected to produce incorrect results.
- --ignoreForNormalization, -ignore
A list of space-delimited chromosome names containing those chromosomes that should be excluded for computing the normalization. This is useful when considering samples with unequal coverage across chromosomes, like male samples. An usage examples is –ignoreForNormalization chrX chrM.
- --skipNonCoveredRegions, --skipNAs
This parameter determines if non-covered regions (regions without overlapping reads) in a BAM file should be skipped. The default is to treat those regions as having a value of zero. The decision to skip non-covered regions depends on the interpretation of the data. Non-covered regions may represent, for example, repetitive regions that should be skipped.
- --smoothLength
The smooth length defines a window, larger than the binSize, to average the number of reads. For example, if the –binSize is set to 20 and the –smoothLength is set to 60, then, for each bin, the average of the bin and its left and right neighbors is considered. Any value smaller than –binSize will be ignored and no smoothing will be applied.
Read processing options
- --extendReads, -e
This parameter allows the extension of reads to fragment size. If set, each read is extended, without exception. NOTE: This feature is generally NOT recommended for spliced-read data, such as RNA-seq, as it would extend reads over skipped regions. Single-end: Requires a user specified value for the final fragment length. Reads that already exceed this fragment length will not be extended. Paired-end: Reads with mates are always extended to match the fragment size defined by the two read mates. Unmated reads, mate reads that map too far apart (>4x fragment length) or even map to different chromosomes are treated like single-end reads. The input of a fragment length value is optional. If no value is specified, it is estimated from the data (mean of the fragment size of all mate reads).
- --ignoreDuplicates
If set, reads that have the same orientation and start position will be considered only once. If reads are paired, the mate’s position also has to coincide to ignore a read.
- --minMappingQuality
If set, only reads that have a mapping quality score of at least this are considered.
- --centerReads
By adding this option, reads are centered with respect to the fragment length. For paired-end data, the read is centered at the fragment length defined by the two ends of the fragment. For single-end data, the given fragment length is used. This option is useful to get a sharper signal around enriched regions.
- --samFlagInclude
Include reads based on the SAM flag. For example, to get only reads that are the first mate, use a flag of 64. This is useful to count properly paired reads only once, as otherwise the second mate will be also considered for the coverage. (Default: None)
- --samFlagExclude
Exclude reads based on the SAM flag. For example, to get only reads that map to the forward strand, use –samFlagExclude 16, where 16 is the SAM flag for reads that map to the reverse strand. (Default: None)
- --minFragmentLength
The minimum fragment length needed for read/pair inclusion. This option is primarily useful in ATACseq experiments, for filtering mono- or di-nucleosome fragments. (Default: 0)
- --maxFragmentLength
The maximum fragment length needed for read/pair inclusion. (Default: 0)
Usage hints
A smaller bin size value will result in a higher resolution of the coverage track but also in a larger file size.
The 1x normalization (RPGC) requires the input of a value for the effective genome size, which is the mappable part of the reference genome. Of course, this value is species-specific. The command line help of this tool offers suggestions for a number of model species.
It might be useful for some studies to exclude certain chromosomes in order to avoid biases, e.g. chromosome X, as male mice contain a pair of each autosome, but usually only a single X chromosome.
By default, the read length is NOT extended! This is the preferred setting for spliced-read data like RNA-seq, where one usually wants to rely on the detected read locations only. A read extension would neglect potential splice sites in the unmapped part of the fragment. Other data, e.g. Chip-seq, where fragments are known to map contiguously, should be processed with read extension (
--extendReads [INTEGER]).For paired-end data, the fragment length is generally defined by the two read mates. The user provided fragment length is only used as a fallback for singletons or mate reads that map too far apart (with a distance greater than four times the fragment length or are located on different chromosomes).
Warning
If you already normalized for GC bias using correctGCbias, you should absolutely NOT set the parameter --ignoreDuplicates!
Note
Like BAM files, bigWig files are compressed, binary files. If you would like to see the coverage values, choose the bedGraph output via --outFileFormat.
Usage example for ChIP-seq
This is an example for ChIP-seq data using additional options (smaller bin size for higher resolution, normalizing coverage to 1x mouse genome size, excluding chromosome X during the normalization step, and extending reads):
bamCoverage --bam a.bam -o a.SeqDepthNorm.bw \
--binSize 10
--normalizeUsing RPGC
--effectiveGenomeSize 2150570000
--ignoreForNormalization chrX
--extendReads
If you had run the command with --outFileFormat bedgraph, you could easily peak into the resulting file.
$ head SeqDepthNorm_chr19.bedgraph
19 60150 60250 9.32
19 60250 60450 18.65
19 60450 60650 27.97
19 60650 60950 37.29
19 60950 61000 27.97
19 61000 61050 18.65
19 61050 61150 27.97
19 61150 61200 18.65
19 61200 61300 9.32
19 61300 61350 18.65
As you can see, each row corresponds to one region. If consecutive bins have the same number of reads overlapping, they will be merged.
Usage examples for RNA-seq
Note that some BAM files are filtered based on SAM flags (Explain SAM flags).
Regular bigWig track
bamCoverage -b a.bam -o a.bw
Separate tracks for each strand
Sometimes it makes sense to generate two independent bigWig files for all reads on the forward and reverse strand, respectively.
As of deepTools version 2.2, one can simply use the --filterRNAstrand option, such as --filterRNAstrand forward or --filterRNAstrand reverse.
This handles paired-end and single-end datasets. For older versions of deepTools, please see the instructions below.
Note
The --filterRNAstrand option assumes the sequencing library generated from ILLUMINA dUTP/NSR/NNSR methods, which are the most commonly used method for
library preparation, where Read 2 (R2) is in the direction of RNA strand (reverse-stranded library). However other methods exist, which generate read
R1 in the direction of RNA strand (see this review). For these libraries,
--filterRNAstrand will have an opposite behavior, i.e. --filterRNAstrand forward will give you reverse strand signal and vice-versa.
Versions before 2.2
To follow the examples, you need to know that -f will tell samtools view to include reads with the indicated flag, while -F will lead to the exclusion of reads with the respective flag.
For a stranded `single-end` library
# Forward strand
bamCoverage -b a.bam -o a.fwd.bw --samFlagExclude 16
# Reverse strand
bamCoverage -b a.bam -o a.rev.bw --samFlagInclude 16
For a stranded `paired-end` library
Now, this gets a bit cumbersome, but future releases of deepTools will make this more straight-forward. For now, bear with us and perhaps read up on SAM flags, e.g. here.
For paired-end samples, we assume that a proper pair should have the mates on opposing strands where the Illumina strand-specific protocol produces reads in a R2-R1 orientation. We basically follow the recipe given in this biostars tutorial.
To get the file for transcripts that originated from the forward strand:
# include reads that are 2nd in a pair (128);
# exclude reads that are mapped to the reverse strand (16)
$ samtools view -b -f 128 -F 16 a.bam > a.fwd1.bam
# exclude reads that are mapped to the reverse strand (16) and
# first in a pair (64): 64 + 16 = 80
$ samtools view -b -f 80 a.bam > a.fwd2.bam
# combine the temporary files
$ samtools merge -f fwd.bam a.fwd1.bam a.fwd2.bam
# index the filtered BAM file
$ samtools index fwd.bam
# run bamCoverage
$ bamCoverage -b fwd.bam -o a.fwd.bigWig
# remove the temporary files
$ rm a.fwd*.bam
To get the file for transcripts that originated from the reverse strand:
# include reads that map to the reverse strand (128)
# and are second in a pair (16): 128 + 16 = 144
$ samtools view -b -f 144 a.bam > a.rev1.bam
# include reads that are first in a pair (64), but
# exclude those ones that map to the reverse strand (16)
$ samtools view -b -f 64 -F 16 a.bam > a.rev2.bam
# merge the temporary files
$ samtools merge -f rev.bam a.rev1.bam a.rev2.bam
# index the merged, filtered BAM file
$ samtools index rev.bam
# run bamCoverage
$ bamCoverage -b rev.bam -o a.rev.bw
# remove temporary files
$ rm a.rev*.bam