plotHeatmap¶
This tool creates a heatmap for a score associated to genomic regions. The program requires a preprocessed matrix generated by the tool computeMatrix.
usage: plotHeatmap [--matrixFile MATRIXFILE] --outFileName OUTFILENAME
[--outFileSortedRegions FILE] [--outFileNameMatrix FILE]
[--kmeans KMEANS] [--hclust HCLUST] [--help] [--version]
[--sortRegions {descend,ascend,no}]
[--sortUsing {mean,median,max,min,sum,region_length}]
[--averageTypeSummaryPlot {mean,median,min,max,std,sum}]
[--missingDataColor MISSINGDATACOLOR] [--colorMap COLORMAP]
[--colorList COLORLIST [COLORLIST ...]]
[--colorNumber COLORNUMBER] [--zMin ZMIN] [--zMax ZMAX]
[--heatmapHeight HEATMAPHEIGHT]
[--heatmapWidth HEATMAPWIDTH]
[--whatToShow {plot, heatmap and colorbar,plot and heatmap,heatmap only,heatmap and colorbar}]
[--xAxisLabel XAXISLABEL] [--startLabel STARTLABEL]
[--endLabel ENDLABEL] [--refPointLabel REFPOINTLABEL]
[--regionsLabel REGIONSLABEL]
[--samplesLabel SAMPLESLABEL [SAMPLESLABEL ...]]
[--plotTitle PLOTTITLE] [--yAxisLabel YAXISLABEL]
[--yMin YMIN] [--yMax YMAX]
[--legendLocation {best,upper-right,upper-left,upper-center,lower-left,lower-right,lower-center,center,center-left,center-right,none}]
[--perGroup] [--plotFileFormat] [--verbose]
- Required arguments
--matrixFile, -m Matrix file from the computeMatrix tool. --outFileName, -out File name to save the image to. The file ending will be used to determine the image format. The available options are: “png”, “eps”, “pdf” and “svg”, e.g., MyHeatmap.png. - Output options
--outFileSortedRegions File name into which the regions are saved after skipping zeros or min/max threshold values. The order of the regions in the file follows the sorting order selected. This is useful, for example, to generate other heatmaps while keeping the sorting of the first heatmap. Example: Heatmap1sortedRegions.bed --outFileNameMatrix If this option is given, then the matrix of values underlying the heatmap will be saved using this name, e.g. MyMatrix.tab. - Clustering arguments
--kmeans Number of clusters to compute. When this option is set, the matrix is split into clusters using the k-means algorithm. Only works for data that is not grouped, otherwise only the first group will be clustered. If more specific clustering methods are required, then save the underlying matrix and run the clustering using other software. The plotting of the clustering may fail with an error if a cluster has very few members compared to the total number or regions. --hclust Number of clusters to compute. When this option is set, then the matrix is split into clusters using the hierarchical clustering algorithm, using “ward linkage”. Only works for data that is not grouped, otherwise only the first group will be clustered. –hclust could be very slow if you have >1000 regions. In those cases, you might prefer –kmeans or if more clustering methods are required you can save the underlying matrix and run the clustering using other software. The plotting of the clustering may fail with an error if a cluster has very few members compared to the total number of regions. - Optional arguments
--version show program’s version number and exit --sortRegions=descend Whether the heatmap should present the regions sorted. The default is to sort in descending order based on the mean value per region.
Possible choices: descend, ascend, no
--sortUsing=mean Indicate which method should be used for sorting. For each row the method is computed.
Possible choices: mean, median, max, min, sum, region_length
--averageTypeSummaryPlot=mean Define the type of statistic that should be plotted in the summary image above the heatmap. The options are: “mean”, “median”, “min”, “max”, “sum” and “std”.
Possible choices: mean, median, min, max, std, sum
--missingDataColor=black If –missingDataAsZero is not set, such cases will be colored in black by default. Using this parameter, a different color can be set. A value between 0 and 1 will be used for a gray scale (black is 0). For a list of possible color names see: http://packages.python.org/ete2/reference/reference_svgcolors.html. Other colors can be specified using the #rrggbb notation. --colorMap=RdYlBu Color map to use for the heatmap. Available values can be seen here: http://www.astro.lsa.umich.edu/~msshin/science/code/matplotlib_cm/ The available options are: ‘Spectral’, ‘summer’, ‘coolwarm’, ‘Set1’, ‘Set2’, ‘Set3’, ‘Dark2’, ‘hot’, ‘RdPu’, ‘YlGnBu’, ‘RdYlBu’, ‘gist_stern’, ‘cool’, ‘gray’, ‘GnBu’, ‘gist_ncar’, ‘gist_rainbow’, ‘CMRmap’, ‘bone’, ‘RdYlGn’, ‘spring’, ‘terrain’, ‘PuBu’, ‘spectral’, ‘gist_yarg’, ‘BuGn’, ‘bwr’, ‘cubehelix’, ‘YlOrRd’, ‘Greens’, ‘PRGn’, ‘gist_heat’, ‘Paired’, ‘hsv’, ‘Pastel2’, ‘Pastel1’, ‘BuPu’, ‘copper’, ‘OrRd’, ‘brg’, ‘gnuplot2’, ‘jet’, ‘gist_earth’, ‘Oranges’, ‘PiYG’, ‘YlGn’, ‘Accent’, ‘gist_gray’, ‘flag’, ‘BrBG’, ‘Reds’, ‘RdGy’, ‘PuRd’, ‘Blues’, ‘Greys’, ‘autumn’, ‘pink’, ‘binary’, ‘winter’, ‘gnuplot’, ‘RdBu’, ‘prism’, ‘YlOrBr’, ‘rainbow’, ‘seismic’, ‘Purples’, ‘ocean’, ‘PuOr’, ‘PuBuGn’, ‘nipy_spectral’, ‘afmhot’ --colorList List of colors to use to create a colormap. For example, if –colorList black yellow blue is set (colors separated by spaces) then a color map that starts with black, continues to yellow and finishes in blue is created. If this option isselected, it overrides the –colorMap selected. --colorNumber=256 N.B., –colorList is required for an effect. This controls the number of transitions from one color to the other. If –colorNumber is the number of colors in –colorList then there will be no transitions between the colors. --zMin, -min Minimum value for the heatmap intensities. --zMax, -max Maximum value for the heatmap intensities. --heatmapHeight=28 Plot height in cm. The default for the heatmap height is 25. The minimum value is 3 and the maximum is 100. --heatmapWidth=4 Plot width in cm. The default value is 4 The minimum value is 1 and the maximum is 100. --whatToShow=plot, heatmap and colorbar The default is to include a summary or profile plot on top of the heatmap and a heatmap colorbar. Other options are: “plot and heatmap”, “heatmap only”, “heatmap and colorbar”, and the default “plot, heatmap and colorbar”.
Possible choices: plot, heatmap and colorbar, plot and heatmap, heatmap only, heatmap and colorbar
--xAxisLabel=gene distance (bp), -x=gene distance (bp) Description for the x-axis label. --startLabel=TSS [only for scale-regions mode] Label shown in the plot for the start of the region. Default is TSS (transcription start site), but could be changed to anything, e.g. “peak start”. Same for the –endLabel option. See below. --endLabel=TES [only for scale-regions mode] Label shown in the plot for the region end. Default is TES (transcription end site). --refPointLabel=TSS [only for reference-point mode] Label shown in the plot for the reference-point. Default is the same as the reference point selected (e.g. TSS), but could be anything, e.g. “peak start”. --regionsLabel=genes, -z=genes Labels for the regions plotted in the heatmap. If more than one region is being plotted a list of labels separated by comma and limited by quotes, is required. For example, –regionsLabel “label1, label2”. Default is “genes”. --samplesLabel Labels for the samples plotted. The default is to use the file name of the sample. The sample names should be separated by spaces and quoted if a label itselfcontains a space E.g. –samplesLabel label-1 “label 2” --plotTitle=, -T= Title of the plot, to be printed on top of the generated image. Leave blank for no title. --yAxisLabel=, -y= Y-axis label for the top panel. --yMin Minimum value for the Y-axis. --yMax Maximum value for the Y-axis. --legendLocation=best Location for the legend in the summary plot. Note that “none” does not work for the profiler.
Possible choices: best, upper-right, upper-left, upper-center, lower-left, lower-right, lower-center, center, center-left, center-right, none
--perGroup=False The default is to plot all groups of regions by sample. Using this option instead plots all samples by group of regions. Note that this is only useful if you have multiple groups of regions. by sample rather than group. --plotFileFormat Image format type. If given, this option overrides the image format based on the plotFile ending. The available options are: “png”, “eps”, “pdf” and “svg”
Possible choices: png, pdf, svg, eps
--verbose=False If set, warning messages and additional information are given.
An example usage is: plotHeatmap -m <matrix file>
Usage Example:¶
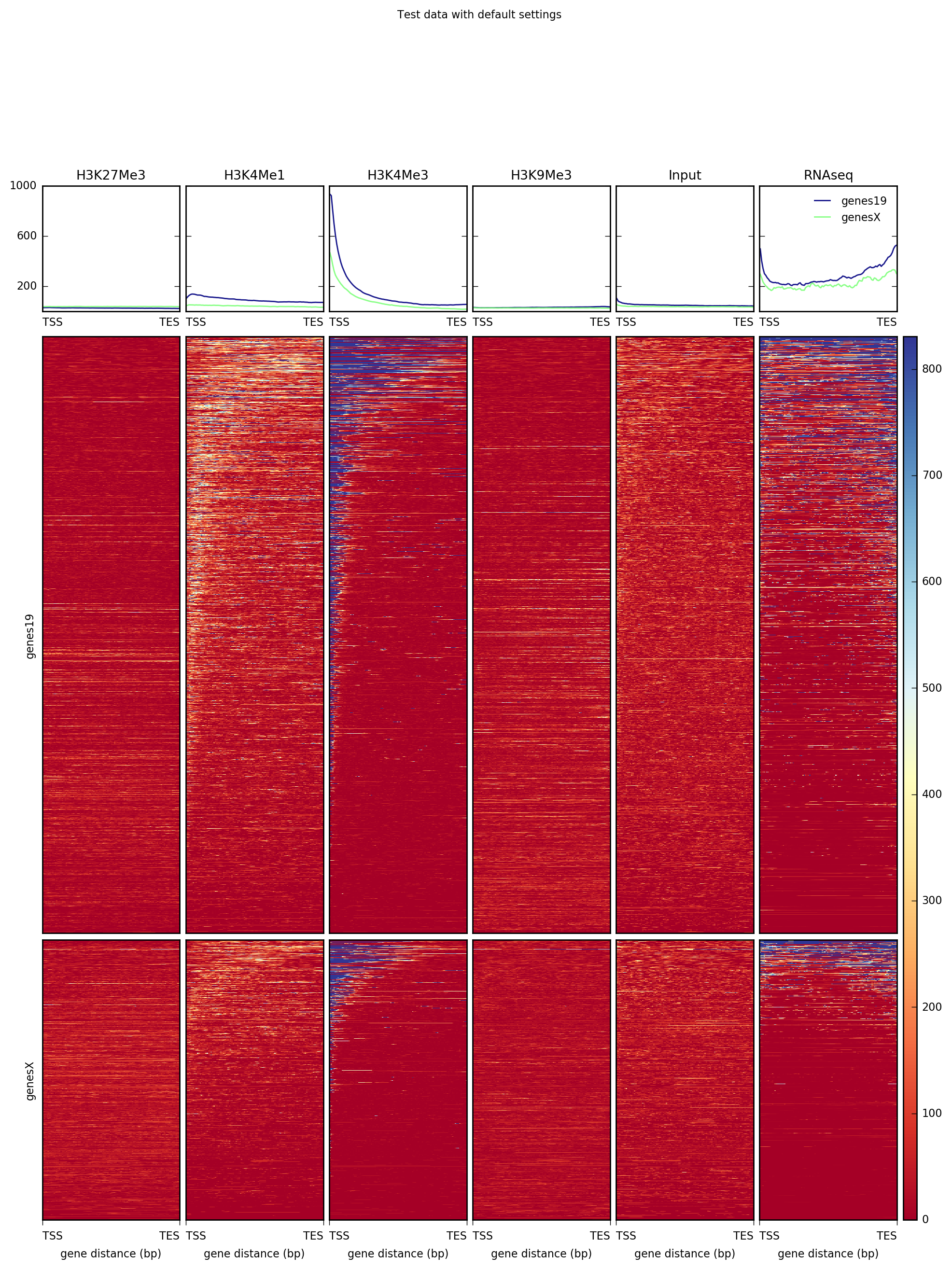
The following example creates a heatmap over hg19 transcripts for our test ENCODE datasets. Note that the matrix contains multiple groups of regions (in this case, one for each chromosome used).
plotHeatmap -m matrix_two_groups.gz \
-out ExampleHeatmap1.png \
--plotTitle "Test data with default settings"
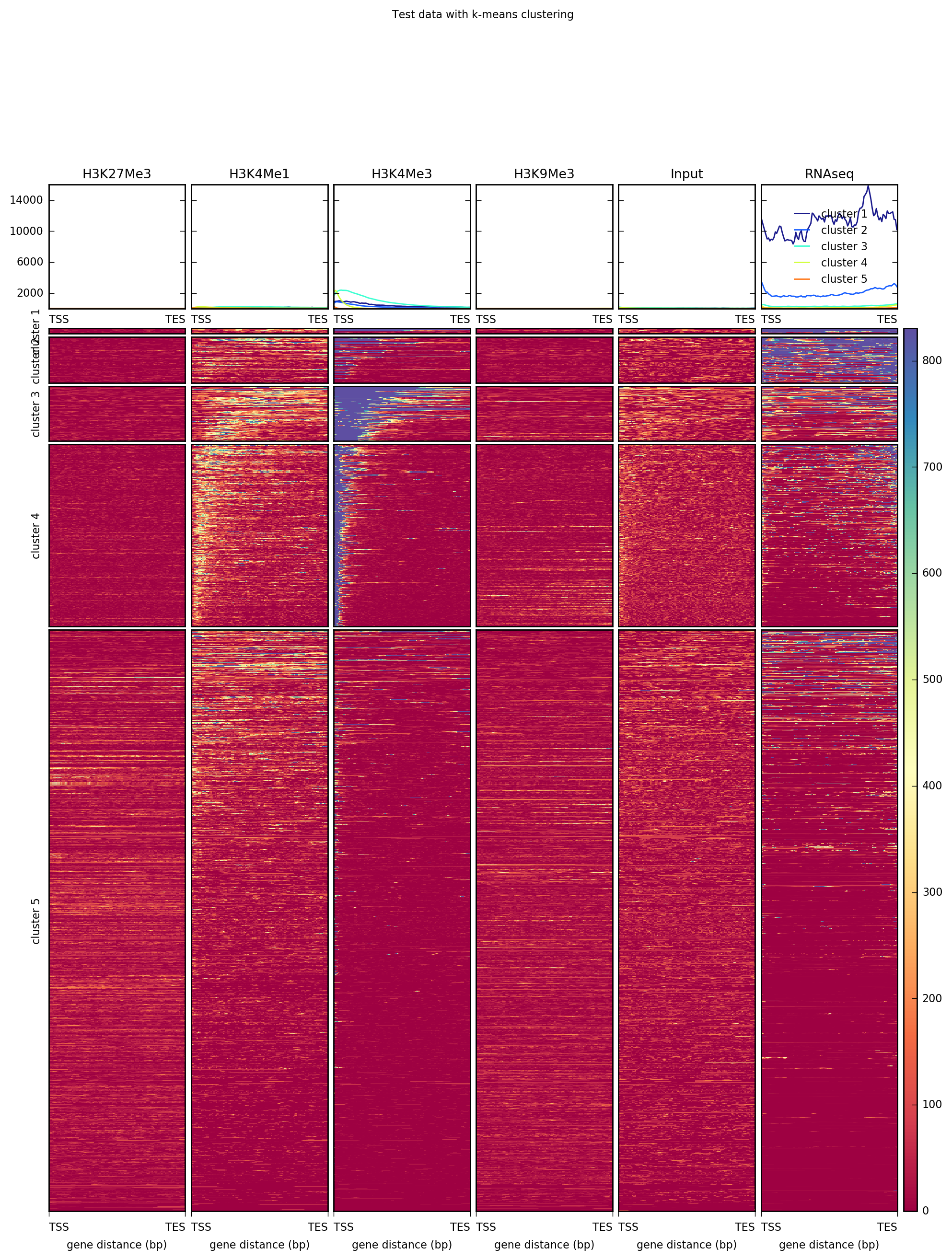
plotHeatmap has many options, including the ability to do k-means clustering and change the color map.
plotHeatmap -m matrix_two_groups.gz \
-out ExampleHeatmap2.png \
--colorMap Spectral \
--kmeans 5 \
--plotTitle "Test data with k-means clustering"