bamPEFragmentSize¶
This tool calculates the fragment sizes for read pairs given a BAM file from paired-end sequencing.Several regions are sampled depending on the size of the genome and number of processors to estimate thesummary statistics on the fragment lengths. Properly paired reads are preferred for computation, i.e., it will only use discordant pairs if no concordant alignments overlap with a given region. The default setting simply prints the summary statistics to the screen.
usage: bamPEFragmentSize [-h] [--histogram FILE] [--numberOfProcessors INT]
[--plotTitle PLOTTITLE] [--binSize INT]
[--distanceBetweenBins INT] [--verbose] [--version]
bam-file
- Positional arguments:
bam BAM file to process - optional arguments
--histogram, -hist Save a .png file with a histogram of the fragment length distribution. --numberOfProcessors=1, -p=1 Number of processors to use. The default is to use 1. --plotTitle=, -T= Title of the plot, to be printed on top of the generated image. Leave blank for no title. --binSize=1000, -bs=1000 Length in bases of the window used to sample the genome. (default 1000) --distanceBetweenBins=1000000, -n=1000000 To reduce the computation time, not every possible genomic bin is sampled. This option allows you to set the distance between bins actually sampled from. Larger numbers are sufficient for high coverage samples, while smaller values are useful for lower coverage samples. Note that if you specify a value that results in too few (<1000) reads sampled, the value will be decreased. (default 1000000) --verbose=False Set if processing data messages are wanted. --version show program’s version number and exit
Note
This tool accepts only one BAM file at a time.
Example usage¶
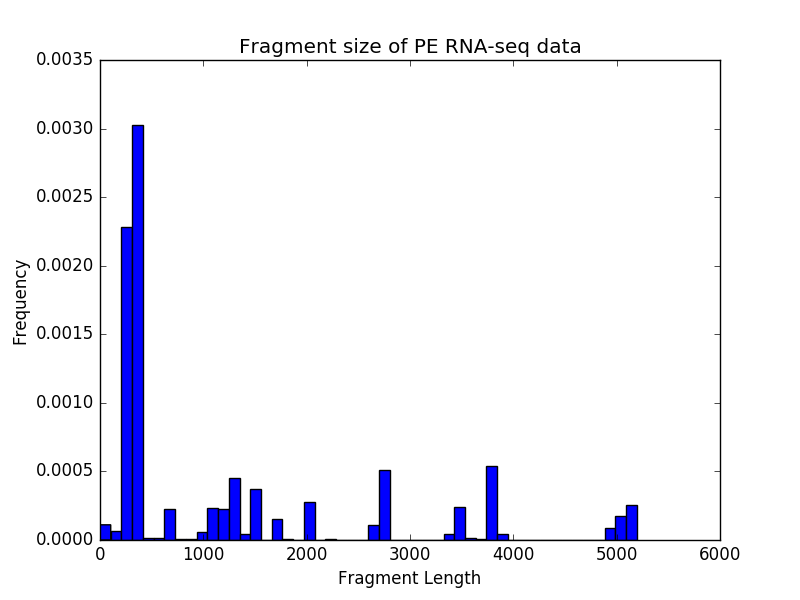
$ deepTools2.0/bin/bamPEFragmentSize \
-hist fragmentSize.png \
-T "Fragment size of PE RNA-seq data" \
testFiles/RNAseq.bam
Sample size: 12850
Fragment lengths:
Min.: 0.0
1st Qu.: 313.0
Mean: 2597.03237354
Median: 357.0
3rd Qu.: 2726.0
Max.: 384622.0
Std: 7066.11863701
Read lengths:
Min.: 20.0
1st Qu.: 101.0
Mean: 99.4182101167
Median: 101.0
3rd Qu.: 101.0
Max.: 101.0
Std: 7.64455778462
| deepTools Galaxy. | code @ github. |