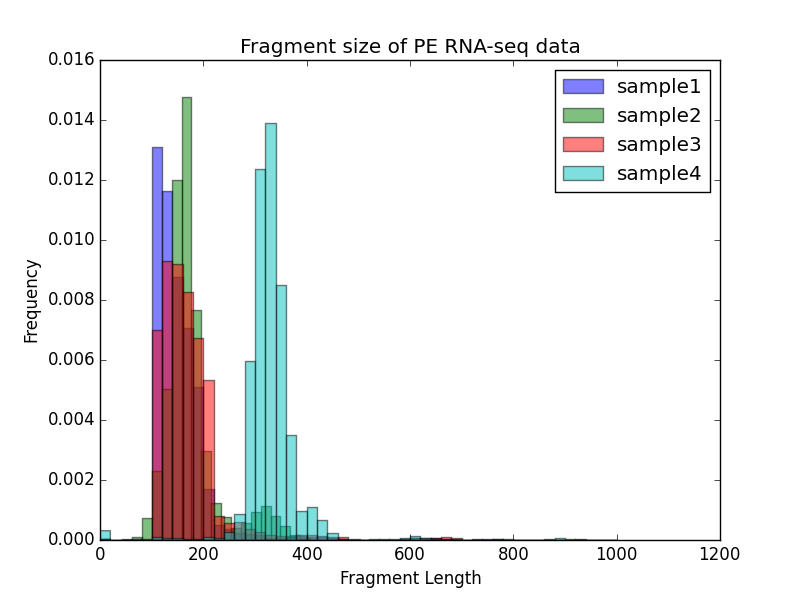
bamPEFragmentSize¶
Example usage¶
$ deepTools2.0/bin/bamPEFragmentSize \
-hist fragmentSize.png \
-T "Fragment size of PE RNA-seq data" \
--maxFragmentLength 1000 \
-b testFiles/RNAseq_sample1.bam testFiles/RNAseq_sample2.bam \
testFiles/RNAseq_sample3.bam testFiles/RNAseq_sample4.bam \
-samplesLabel sample1 sample2 sample3 sample4
## Output
BAM file : testFiles/RNAseq_sample1.bam
Sample size: 10815
Fragment lengths:
Min.: 0.0
1st Qu.: 311.0
Mean: 8960.68987517
Median: 331.0
3rd Qu.: 362.0
Max.: 53574842.0
Std: 572421.46625
Read lengths:
Min.: 20.0
1st Qu.: 101.0
Mean: 99.1621821544
Median: 101.0
3rd Qu.: 101.0
Max.: 101.0
Std: 9.16567362755
BAM file : testFiles/RNAseq_sample2.bam
Sample size: 6771
Fragment lengths:
Min.: 43.0
1st Qu.: 148.0
Mean: 176.465071629
Median: 164.0
3rd Qu.: 185.0
Max.: 500.0
Std: 53.733877263
......(output truncated)
If the --table option is specified, the summary statistics are additionally printed in a tabular format:
Frag. Len. Min. Frag. Len. 1st. Qu. Frag. Len. Mean Frag. Len. Median Frag. Len. 3rd Qu. Frag. Len. Max Frag. Len. Std. Read Len. Min. Read Len. 1st. Qu. Read Len. Mean Read Len. Median Read Len. 3rd Qu. Read Len. Max Read Len. Std.
bowtie2 test1.bam 241.0 241.5 244.666666667 242.0 246.5 251.0 4.49691252108 251.0 251.0 251.0 251.0 251.0 251.0 0.0
If the --outRawFragmentLengths option is provided, another history item will be produced, containing the raw data underlying the histogram. It has the following format:
#bamPEFragmentSize
Size Occurrences Sample
241 1 bowtie2 test1.bam
242 1 bowtie2 test1.bam
251 1 bowtie2 test1.bam
The “Size” is the fragment (or read, for single-end datasets) size and “Occurrences” are the number of times reads/fragments with that length were observed. For easing downstream processing, the sample name is a lso included on each row.
| deepTools Galaxy. | code @ github. |