plotFingerprint¶
This quality control will most likely be of interest for you if you are dealing with ChIP-seq samples as a pressing question in ChIP-seq experiments is “Did my ChIP work?”, i.e. did the antibody-treatment enrich sufficiently so that the ChIP signal can be separated from the background signal? (After all, around 90% of all DNA fragments in a ChIP experiment will represent the genomic background).
Note
We’ve termed the plots described here “fingerprints” because we feel that they help us judging individual ChIP-seq files, but the original idea came from Diaz et al.
This tool samples indexed BAM files and plots a profile of cumulative read coverages for each. All reads overlapping a window (bin) of the specified length are counted; these counts are sorted and the cumulative sum is finally plotted.
usage: An example usage is: plotFingerprint -b treatment.bam control.bam -plot fingerprint.png
Required arguments¶
| --bamfiles, -b | List of indexed BAM files |
Output¶
| --plotFile, -plot, -o | |
| File name of the output figure. The file ending will be used to determine the image format. The available options are typically: “png”, “eps”, “pdf” and “svg”, e.g. : fingerprint.png. | |
| --outRawCounts | Output file name to save the read counts per bin. |
Read processing options¶
| --extendReads, -e | |
| This parameter allows the extension of reads to fragment size. If set, each read is extended, without exception. NOTE: This feature is generally NOT recommended for spliced-read data, such as RNA-seq, as it would extend reads over skipped regions. Single-end: Requires a user specified value for the final fragment length. Reads that already exceed this fragment length will not be extended. Paired-end: Reads with mates are always extended to match the fragment size defined by the two read mates. Unmated reads, mate reads that map too far apart (>4x fragment length) or even map to different chromosomes are treated like single-end reads. The input of a fragment length value is optional. If no value is specified, it is estimated from the data (mean of the fragment size of all mate reads). | |
| --ignoreDuplicates | |
| If set, reads that have the same orientation and start position will be considered only once. If reads are paired, the mate’s position also has to coincide to ignore a read. | |
| --minMappingQuality | |
| If set, only reads that have a mapping quality score of at least this are considered. | |
| --centerReads | By adding this option, reads are centered with respect to the fragment length. For paired-end data, the read is centered at the fragment length defined by the two ends of the fragment. For single-end data, the given fragment length is used. This option is useful to get a sharper signal around enriched regions. |
| --samFlagInclude | |
| Include reads based on the SAM flag. For example, to get only reads that are the first mate, use a flag of 64. This is useful to count properly paired reads only once, as otherwise the second mate will be also considered for the coverage. | |
| --samFlagExclude | |
| Exclude reads based on the SAM flag. For example, to get only reads that map to the forward strand, use –samFlagExclude 16, where 16 is the SAM flag for reads that map to the reverse strand. | |
| --minFragmentLength | |
| The minimum fragment length needed for read/pair inclusion. This option is primarily useful in ATACseq experiments, for filtering mono- or di-nucleosome fragments. | |
| --maxFragmentLength | |
| The maximum fragment length needed for read/pair inclusion. | |
Optional arguments¶
| --labels, -l | List of labels to use in the output. If not given, the file names will be used instead. Separate the labels by spaces. |
| --smartLabels | Instead of manually specifying labels for the input BAM/bigWig files, this causes deepTools to use the file name after removing the path and extension. |
| --binSize, -bs | Window size in base pairs to sample the genome. This times –numberOfSamples should be less than the genome size. |
| --numberOfSamples, -n | |
| The number of bins that are sampled from the genome, for which the overlapping number of reads is computed. | |
| --plotFileFormat | |
Possible choices: png, pdf, svg, eps, plotly image format type. If given, this option overrides the image format based on the ending given via –plotFile ending. The available options are: “png”, “eps”, “pdf”, “plotly” and “svg” | |
| --plotTitle, -T | |
| Title of the plot, to be printed on top of the generated image. Leave blank for no title. | |
| --skipZeros | If set, then regions with zero overlapping readsfor all given BAM files are ignored. This will result in a reduced number of read counts than that specified in –numberOfSamples |
| --outQualityMetrics | |
| Quality metrics can optionally be output to this file. The file will have one row per input BAM file and columns containing a number of metrics. Please see the online documentation for a longer explanation: http://deeptools.readthedocs.io/en/latest/content/feature/plotFingerprint_QC_metrics.html . | |
| --JSDsample | Reference sample against which to compute the Jensen-Shannon distance and the CHANCE statistics. If this is not specified, then these will not be calculated. If –outQualityMetrics is not specified then this will be ignored. The Jensen-Shannon implementation is based on code from Sitanshu Gakkhar at BCGSC. The CHANCE implementation is based on code from Matthias Haimel. |
| --version | show program’s version number and exit |
| --region, -r | Region of the genome to limit the operation to - this is useful when testing parameters to reduce the computing time. The format is chr:start:end, for example –region chr10 or –region chr10:456700:891000. |
| --blackListFileName, -bl | |
| A BED or GTF file containing regions that should be excluded from all analyses. Currently this works by rejecting genomic chunks that happen to overlap an entry. Consequently, for BAM files, if a read partially overlaps a blacklisted region or a fragment spans over it, then the read/fragment might still be considered. Please note that you should adjust the effective genome size, if relevant. | |
| --numberOfProcessors, -p | |
| Number of processors to use. Type “max/2” to use half the maximum number of processors or “max” to use all available processors. | |
| --verbose, -v | Set to see processing messages. |
Background¶
This tool is based on a method developed by Diaz et al.. It determines how well the signal in the ChIP-seq sample can be differentiated from the background distribution of reads in the control sample. For factors that will enrich well-defined, rather narrow regions (e.g. transcription factors such as p300), the resulting plot can be used to assess the strength of a ChIP, but the broader the enrichments are to be expected, the less clear the plot will be. Vice versa, if you do not know what kind of signal to expect, the fingerprint plot will give you a straight-forward indication of how careful you will have to be during your downstream analyses to separate biological noise from meaningful signal.
Similar to multiBamSummary, plotFingerprint randomly samples genome regions (bins) of a specified length and sums the per-base coverage in indexed [BAM][] (or bigWig) files that overlap with those regions.
These values are then sorted according to their rank and the cumulative sum of read counts is plotted.
What the plots tell you¶
An ideal [input][] with perfect uniform distribution of reads along the genome (i.e. without enrichments in open chromatin etc.) and infinite sequencing coverage should generate a straight diagonal line. A very specific and strong ChIP enrichment will be indicated by a prominent and steep rise of the cumulative sum towards the highest rank. This means that a big chunk of reads from the ChIP sample is located in few bins which corresponds to high, narrow enrichments typically seen for transcription factors.
Here you see 3 different fingerprint plots. We chose these examples to show you how the nature of the ChIP signal (narrow and high vs. wide and not extremely high) is reflected in the “fingerprint” plots.
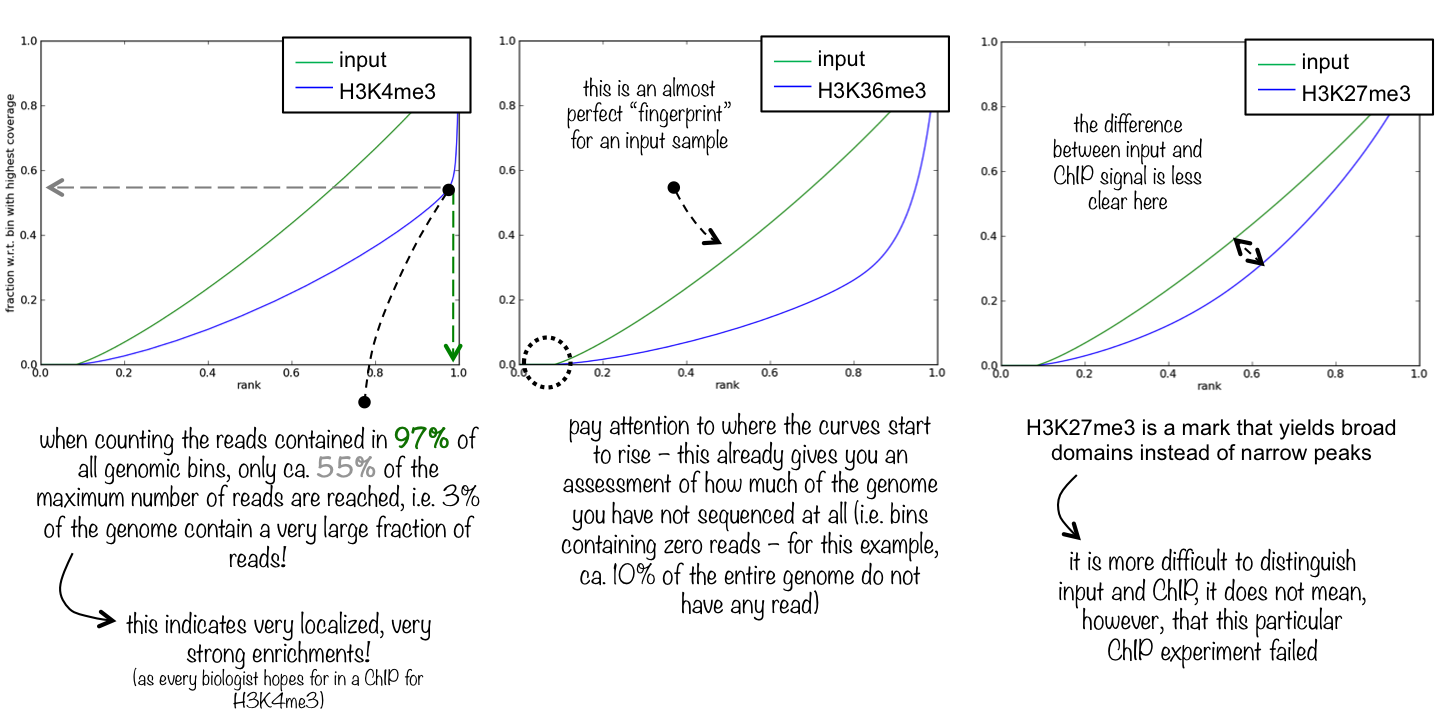
Quality control metrics¶
For a detailed explanation of the QC metrics, please see: plotFingerprint QC metrics.
Usage example¶
The following example generates the fingerprints for the invididual ENCODE histone mark ChIP-seq data sets and their corresponding input (focusing on chromosome 19 and thus adjusting the number of 500 bp bins that are being sampled using --numberOfSamples to avoid overlapping bins).
$ deepTools2.0/bin/plotFingerprint \
-b testFiles/*bam \
--labels H3K27me3 H3K4me1 H3K4me3 H3K9me3 input \
--minMappingQuality 30 --skipZeros \
--region 19 --numberOfSamples 50000 \
-T "Fingerprints of different samples" \
--plotFile fingerprints.png \
--outRawCounts fingerprints.tab
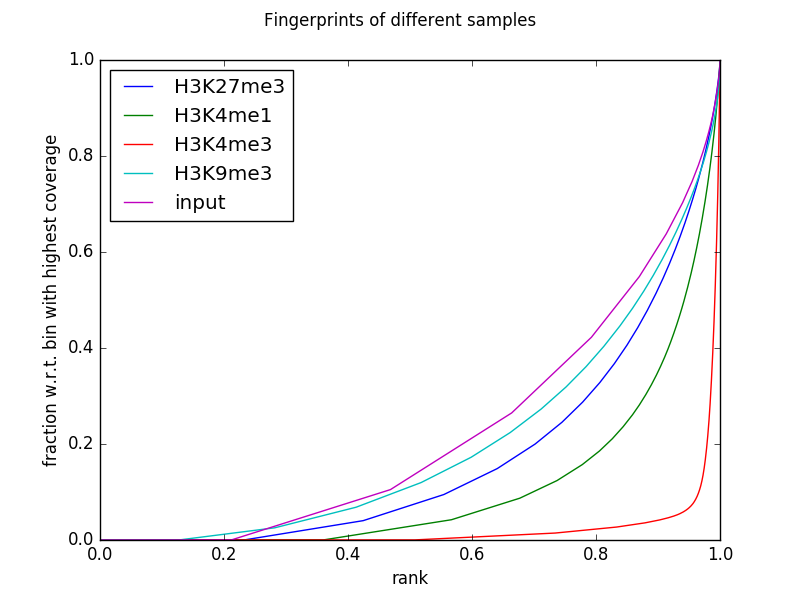
The table that you can obtain via --outRawCounts simply contains the sum of the per-base coverage inside each sampled genome bin. For the plot above, each column is sorted in increasing order and then the cumulative sum is plotted.
$ head fingerprints.tab
#plotFingerprint --outRawCounts
'H3K27me3' 'H3K4me1' 'H3K4me3' 'H3K9me3' 'input'
1 0 0 0 0
0 0 0 0 1
0 1 0 0 0
12 0 0 3 3
3 0 1 1 0
6 4 0 1 0
1 0 0 0 0
4 1 1 1 0
1 0 0 0 0
| deepTools Galaxy. | code @ github. |