plotEnrichment
Tool for calculating and plotting the signal enrichment in either regions in BED format or feature types (column 3) in GTF format. The underlying datapoints can also be output. Metrics are plotted as a fraction of total reads. Regions in a BED file are assigned to the ‘peak’ feature.
detailed help:
plotEnrichment -h
usage: plotEnrichment -b sample1.bam sample2.bam --BED peaks.bed -o enrichment.png
help: plotEnrichment -h / plotEnrichment --help
Required arguments
- --bamfiles, -b
List of indexed bam files separated by spaces.
- --BED
Limits the enrichment analysis to the regions specified in these BED/GTF files. Enrichment is calculated as the number of reads overlapping each feature type. The feature type is column 3 in a GTF file and “peak” for BED files.
Optional arguments
- --plotFile, -o
File to save the plot to. The file extension determines the format, so heatmap.pdf will save the heatmap in PDF format. The available formats are: .png, .eps, .pdf and .svg.
- --attributeKey
Instead of deriving labels from the feature column in a GTF file, use the given attribute key, such as gene_biotype. For BED files or entries without the attribute key, None is used as the label.
- --labels, -l
User defined labels instead of default labels from file names. Multiple labels have to be separated by spaces, e.g. –labels sample1 sample2 sample3
- --smartLabels
Instead of manually specifying labels for the input BAM/BED/GTF files, this causes deepTools to use the file name after removing the path and extension. For BED/GTF files, the eventual region name will be overriden if specified inside the file.
- --regionLabels
For BED files, the label given to its region is the file name, but this can be overridden by providing a custom label. For GTF files this is ignored. Note that if you provide labels, you MUST provide one for each BED/GTF file, even though it will be ignored for GTF files.
- --plotTitle, -T
Title of the plot, to be printed on top of the generated image. Leave blank for no title. (Default: )
- --plotFileFormat
Possible choices: png, pdf, svg, eps, plotly
Image format type. If given, this option overrides the image format based on the plotFile ending. The available options are: png, eps, pdf, plotly and svg.
- --outRawCounts
Save the counts per region to a tab-delimited file.
- --perSample
Group the plots by sample, rather than by feature type (the default).
- --variableScales
By default, the y-axes are always 0-100. This allows the axis range to be restricted.
- --plotHeight
Plot height in cm. (Default: 20)
- --plotWidth
Plot width in cm. The minimum value is 1 cm. (Default: 20)
- --colors
List of colors to use for the plotted lines. Color names and html hex strings (e.g., #eeff22) are accepted. The color names should be space separated. For example, –colors red blue green
- --numPlotsPerRow
Number of plots per row (Default: 4)
- --alpha
The alpha channel (transparency) to use for the bars. The default is 0.9 and values must be between 0 and 1.
- --Offset
Uses this offset inside of each read as the signal. This is useful in cases like RiboSeq or GROseq, where the signal is 12, 15 or 0 bases past the start of the read. This can be paired with the –filterRNAstrand option. Note that negative values indicate offsets from the end of each read. A value of 1 indicates the first base of the alignment (taking alignment orientation into account). Likewise, a value of -1 is the last base of the alignment. An offset of 0 is not permitted. If two values are specified, then they will be used to specify a range of positions. Note that specifying something like –Offset 5 -1 will result in the 5th through last position being used, which is equivalent to trimming 4 bases from the 5-prime end of alignments.
- --version
show program’s version number and exit
- --region, -r
Region of the genome to limit the operation to - this is useful when testing parameters to reduce the computing time. The format is chr:start:end, for example –region chr10 or –region chr10:456700:891000.
- --blackListFileName, -bl
A BED or GTF file containing regions that should be excluded from all analyses. Currently this works by rejecting genomic chunks that happen to overlap an entry. Consequently, for BAM files, if a read partially overlaps a blacklisted region or a fragment spans over it, then the read/fragment might still be considered. Please note that you should adjust the effective genome size, if relevant.
- --numberOfProcessors, -p
Number of processors to use. Type “max/2” to use half the maximum number of processors or “max” to use all available processors. (Default: 1)
- --verbose, -v
Set to see processing messages.
BED12 arguments
- --keepExons
For BED12 files, use each exon as a region, rather than columns 2/3
Read processing options
- --extendReads, -e
This parameter allows the extension of reads to fragment size. If set, each read is extended, without exception. NOTE: This feature is generally NOT recommended for spliced-read data, such as RNA-seq, as it would extend reads over skipped regions. Single-end: Requires a user specified value for the final fragment length. Reads that already exceed this fragment length will not be extended. Paired-end: Reads with mates are always extended to match the fragment size defined by the two read mates. Unmated reads, mate reads that map too far apart (>4x fragment length) or even map to different chromosomes are treated like single-end reads. The input of a fragment length value is optional. If no value is specified, it is estimated from the data (mean of the fragment size of all mate reads).
- --ignoreDuplicates
If set, reads that have the same orientation and start position will be considered only once. If reads are paired, the mate’s position also has to coincide to ignore a read.
- --minMappingQuality
If set, only reads that have a mapping quality score of at least this are considered.
- --centerReads
By adding this option, reads are centered with respect to the fragment length. For paired-end data, the read is centered at the fragment length defined by the two ends of the fragment. For single-end data, the given fragment length is used. This option is useful to get a sharper signal around enriched regions.
- --samFlagInclude
Include reads based on the SAM flag. For example, to get only reads that are the first mate, use a flag of 64. This is useful to count properly paired reads only once, as otherwise the second mate will be also considered for the coverage. (Default: None)
- --samFlagExclude
Exclude reads based on the SAM flag. For example, to get only reads that map to the forward strand, use –samFlagExclude 16, where 16 is the SAM flag for reads that map to the reverse strand. (Default: None)
- --minFragmentLength
The minimum fragment length needed for read/pair inclusion. This option is primarily useful in ATACseq experiments, for filtering mono- or di-nucleosome fragments. (Default: 0)
- --maxFragmentLength
The maximum fragment length needed for read/pair inclusion. (Default: 0)
example usages: plotEnrichment -b file1.bam file2.bam –BED peaks.bed -o enrichment.png
Background
It’s often useful to know what percentage of alignments or fragments overlap one or more groups of regions. For example, “fragment of reads in peaks” or FRiP scores are a common QC metric for ChIPseq data that asks such a question. Another example would be seeing what fraction of RNAseq reads are in exons, or genes, since both of these should be high. plotEnrichment allows efficiently answering these sorts of questions.
Usage example
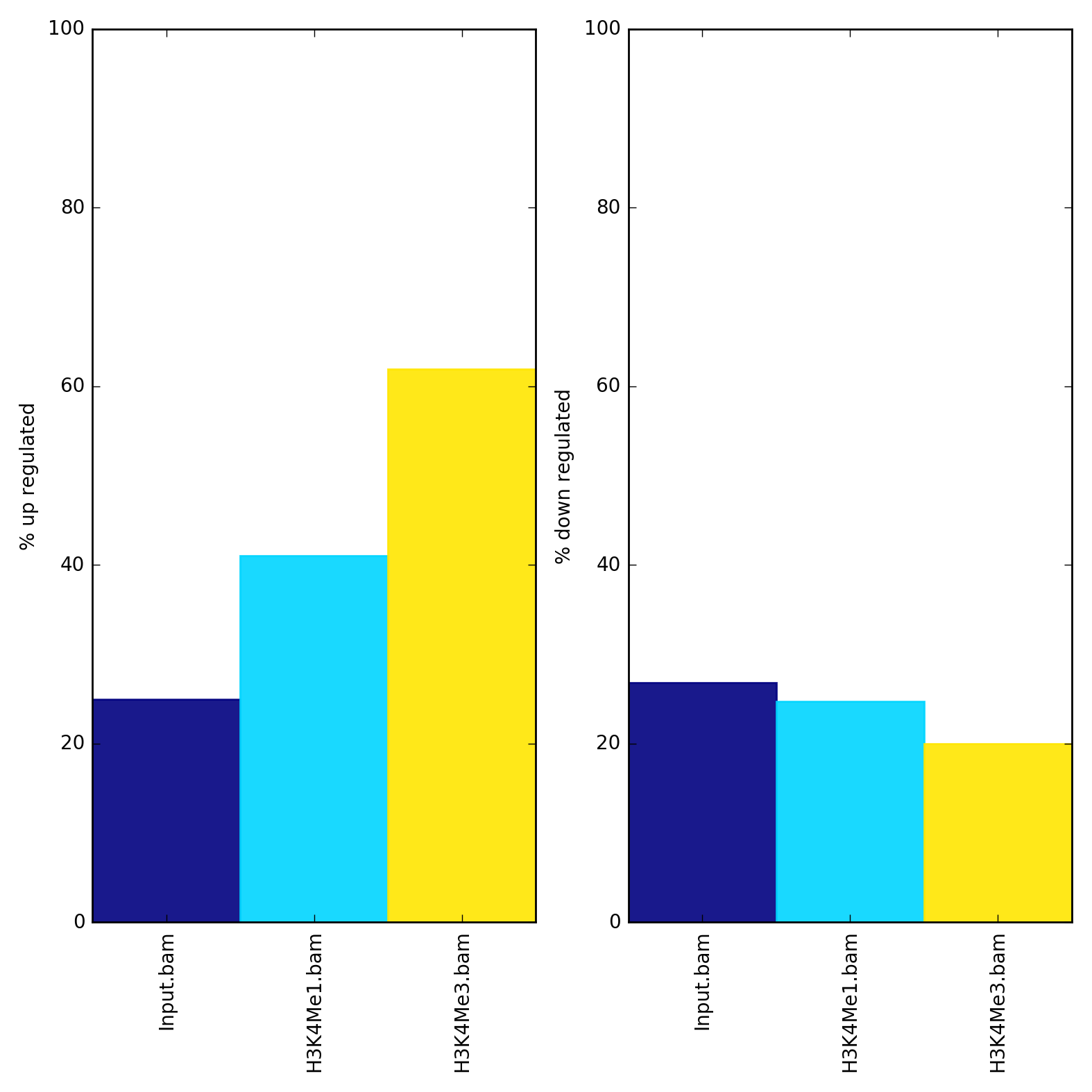
plotEnrichment needs one or more sorted and indexed BAM files and one or more BED and/or GTF files. For GTF files, feature labels are given according to the 3rd column (‘feature’). For BED files, labels are given by the file name, though this can be overriden with the --regionLabels option.
$ plotEnrichment -b Input.bam H3K4Me1.bam H3K4Me3.bam \
--BED up.bed down.bed \
--regionLabels "up regulated" "down regulated" \
-o enrichment.png
The values underlying the plot can also be output with the --outRawCounts option and the y-axis can be auto-adjusted with the --variableScales option.