correctGCBias¶
Hint
For background information about the GC bias assessment and correction, see computeGCBias.
Corrects the GC-bias using Benjamini’s method [Benjamini & Speed (2012). Nucleic acids research, 40(10)]. The tool computeGC bias needs to be run first.
usage: An example usage is:
correctGCBias -b file.bam --effectiveGenomeSize 2150570000 -g mm9.2bit --GCbiasFrequenciesFile freq.txt -o gc_corrected.bam [options]
- Required arguments
--bamfile, -b Sorted BAM file to correct. --effectiveGenomeSize The effective genome size is the portion of the genome that is mappable. Large fractions of the genome are stretches of NNNN that should be discarded. Also, if repetitive regions were not included in the mapping of reads, the effective genome size needs to be adjusted accordingly. Common values are: mm9: 2150570000, hg19:2451960000, dm3:121400000 and ce10:93260000. See Table 2 of http://www.plosone.org/article/info:doi/10.1371/journal.pone.0030377 or http://www.nature.com/nbt/journal/v27/n1/fig_tab/nbt.1518_T1.html for several effective genome sizes. This value is needed to detect enriched regions that, if not discarded, could bias the results. --genome, -g Genome in two bit format. Most genomes can be found here: http://hgdownload.cse.ucsc.edu/gbdb/ Search for the .2bit ending. Otherwise, fasta files can be converted to 2bit using faToTwoBit available here: http://hgdownload.cse.ucsc.edu/admin/exe/ --GCbiasFrequenciesFile, -freq Indicate the output file from computeGCBias containing the observed and expected read frequencies per GC-content. - Output options
--correctedFile, -o Name of the corrected file. The ending will be used to decide the output file format. The options are ”.bam”, ”.bw” for a bigWig file, ”.bg” for a bedGraph file. - Optional arguments
--version show program’s version number and exit --binSize=50, -bs=50 Size of the bins, in bases, for the output of the bigwig/bedgraph file. --region, -r Region of the genome to limit the operation to - this is useful when testing parameters to reduce the computing time. The format is chr:start:end, for example –region chr10 or –region chr10:456700:891000. --numberOfProcessors=max/2, -p=max/2 Number of processors to use. Type “max/2” to use half the maximum number of processors or “max” to use all available processors. --verbose=False, -v=False Set to see processing messages.
Usage example¶
Note
correctGCBias requires the output of computeGCBias.
$ correctGCBias -b H3K27Me3.bam
--effectiveGenomeSize 2695000000
--genome genome.2bit
--GCbiasFrequenciesFile freq_test.txt # output of computeGCBias
-o gc_corrected.bam
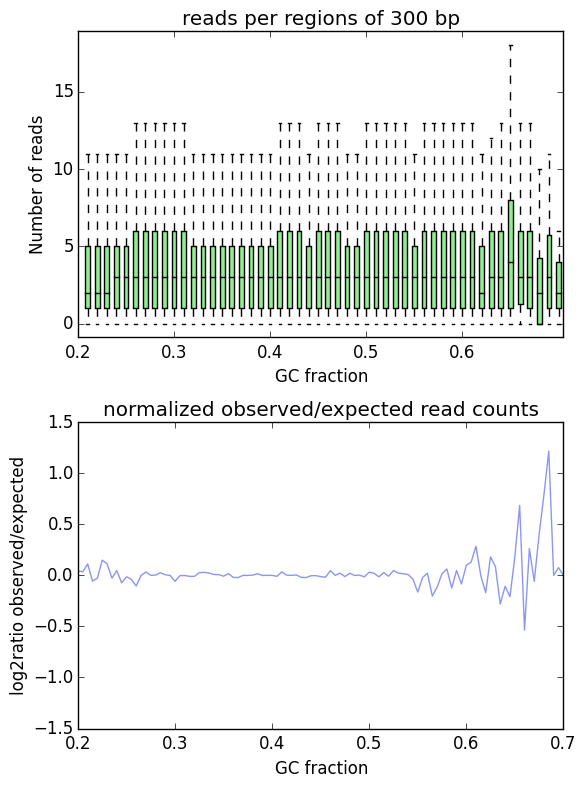
Example output plot¶
The example shows the GC-bias of a corrected BAM file (output from computeGCBias).