deepTools: tools for exploring deep sequencing data
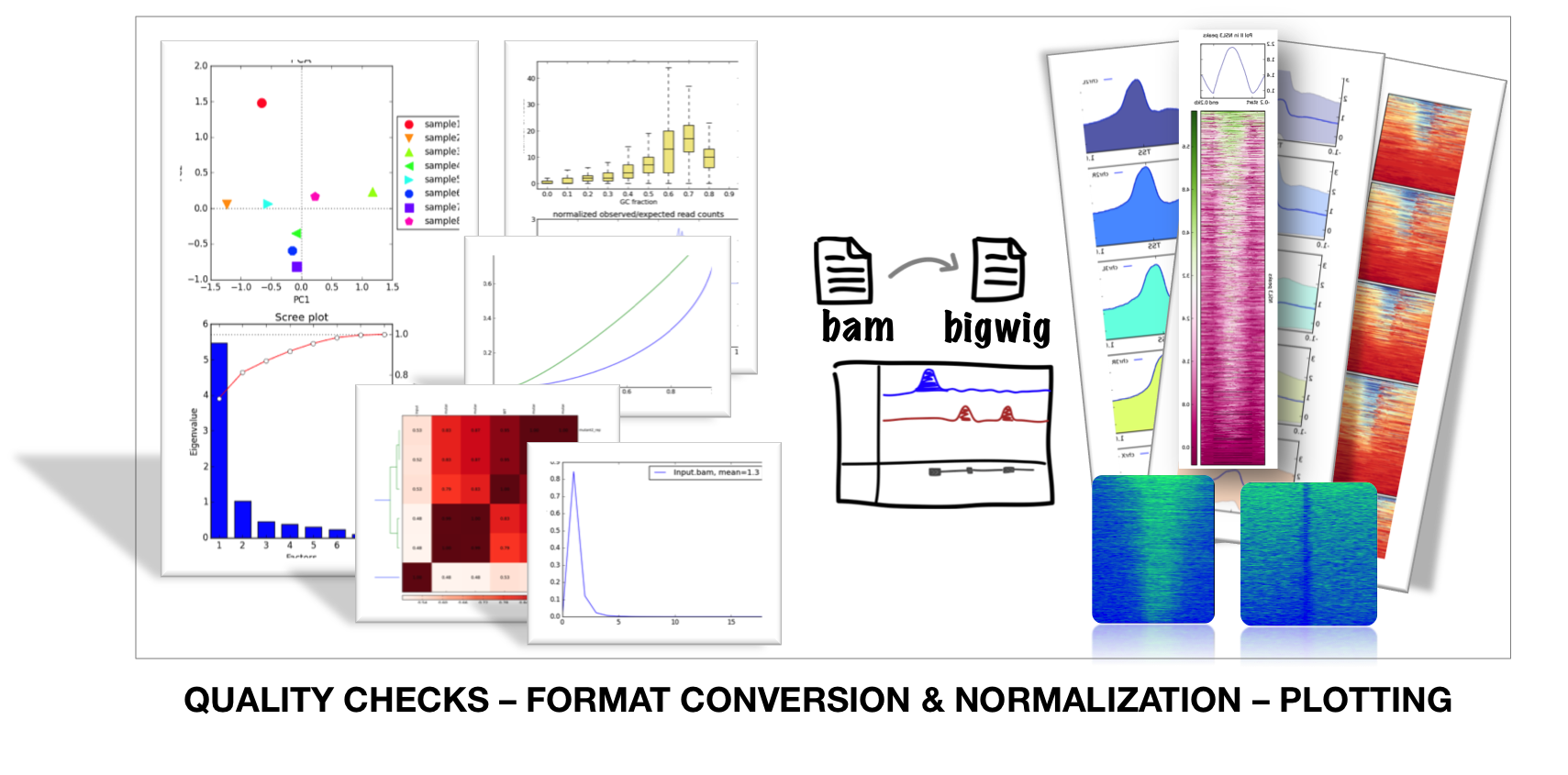
deepTools is a suite of python tools particularly developed for the efficient analysis of high-throughput sequencing data, such as ChIP-seq, RNA-seq or MNase-seq.
There are 3 ways for using deepTools:
Galaxy usage – our public deepTools Galaxy server let’s you use the deepTools within the familiar Galaxy framework without the need to master the command line
command line usage – simply download and install the tools (see Installation and The tools)
API – make use of your favorite deepTools modules in your own python programs (see deepTools API)
The flow chart below depicts the different tool modules that are currently available.
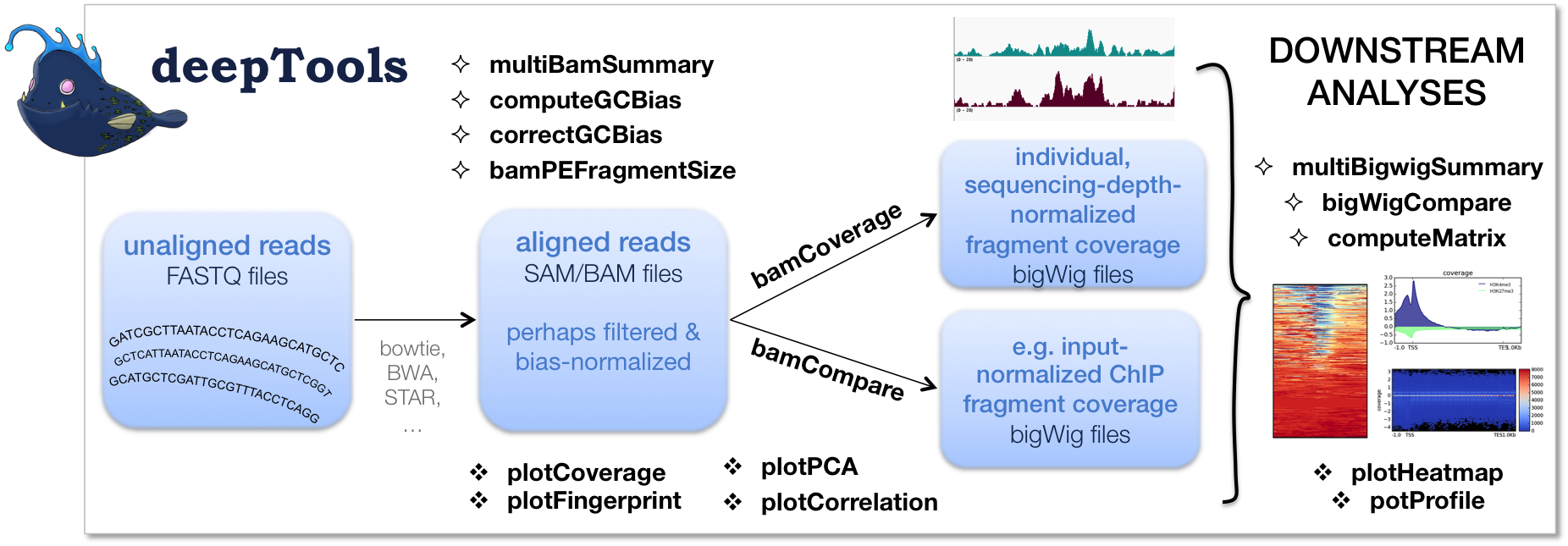
If the file names in the figure mean nothing to you, please make sure to check our Glossary of NGS terms.
Contents:
While developing deepTools, we continuously strive to create software that fulfills the following criteria:
efficiently extract reads from BAM files and perform various computations on them
turn BAM files of aligned reads into bigWig files using different normalization strategies
make use of multiple processors (speed!)
generation of highly customizable images (change colours, size, labels, file format, etc.)
enable customized down-stream analyses, meaning that every data set created can be stored by the user
modular approach - compatibility, flexibility, scalability (i.e. we can add more and more modules and make use of established methods)
Tip
For support or questions please post to Biostars. For bug reports and feature requests please open an issue on github.
Please cite deepTools2 as follows:
Ramírez, Fidel, Devon P. Ryan, Björn Grüning, Vivek Bhardwaj, Fabian Kilpert, Andreas S. Richter, Steffen Heyne, Friederike Dündar, and Thomas Manke. “deepTools2: a next generation web server for deep-sequencing data analysis.” Nucleic Acids Research (2016): gkw257.
This tool suite is developed by the Bioinformatics Facility at the Max Planck Institute for Immunobiology and Epigenetics, Freiburg.