deepTools API example
The following is a short overview of the most useful methods and classes from deepTools. Complete information can be found in the following links: Index and Module Index
Finding read coverage over a region
With deepTools, the read coverage over multiple genomic regions and multiple files can be computed quite quickly using multiple processors.
First, we start with a simple example that is later expanded upon to demonstrate
the use of multipe processors.
In this example we compute the coverage of reads over a small region for bins of 50bp. For this we need the deeptools.countReadsPerBin class.
import deeptools.countReadsPerBin as crpb
We also need a BAM file containing the aligned reads. The BAM file must be indexed to allow quick access to reads falling into the regions of interest.
bam_file = "file.bam"
Now, the CountReadsPerBin object can be initialized.
The first argument to the constructor is a list of BAM files,
which in this case is just one file.
We are going to use a binLength of 50 bases, with subsequent bins adjacent
(i.e., the stepSize between bins is also 50 bases). Overlapping bin
coverages can be used by setting a stepSize smaller than binLength.
cr = crpb.CountReadsPerBin([bam_file], binLength=50, stepSize=50)
Now, we can compute the coverage over a region in chromosome 2 from position 0 to 1000.
cr.count_reads_in_region('chr2L', 0, 1000)
(array([[ 2.],
[ 3.],
[ 1.],
[ 2.],
[ 3.],
[ 2.],
[ 4.],
[ 3.],
[ 2.],
[ 3.],
[ 4.],
[ 6.],
[ 4.],
[ 2.],
[ 2.],
[ 1.]]), '')
The result is a tuple with the first element a numpy array with one row per bin and one column per bam file. Since only one BAM file was used, there is only one column. If a file name for saving the raw data had been specificied, then the temporary file name used for this would appear in the second item of the tuple.
Filtering reads
If reads should be filtered, the relevant options simply need to be passed to the constructor. In the following code, the reads are filtered such that only those with a mapping quality of at least 20 and not aligned to the reverse strand are kept (samFlag_exclude=16, where 16 is the value for reverse reads, see the [SAM Flag Calculator](http://broadinstitute.github.io/picard/explain-flags.html) for more info). Furthermore, duplicated reads are ignored.
cr = crpb.CountReadsPerBin([bam_file], binLength=50, stepSize=50,
minMappingQuality=20,
samFlag_exclude=16,
ignoreDuplicates=True
)
cr.count_reads_in_region('chr2L', 1000000, 1001000)
(array([[ 1.],
[ 1.],
[ 0.],
[ 0.],
[ 0.],
[ 0.],
[ 2.],
[ 3.],
[ 1.],
[ 0.],
[ 1.],
[ 2.],
[ 0.],
[ 0.],
[ 1.],
[ 2.],
[ 1.],
[ 0.],
[ 0.],
[ 0.]]), '')
Sampling the genome
Instead of adjacent bins, as in the previous cases, a genome can simply be sampled. This is useful to estimate some values, like depth of sequencing, without having to look at the complete genome. In the following example, 10,000 positions of size 1 base are going to be queried from three bam files to compute the average depth of sequencing. For this, we set the numberOfSamples parameter in the object constructor.
The run() method is used instead of count_reads_in_region to provide efficient sampling over the entire genome.
cr = crpb.CountReadsPerBin([bam_file1, bam_file2, bam_file3],
binLength=1, numberOfSamples=10000,
numberOfProcessors=10)
sequencing_depth = cr.run()
print sequencing_depth.mean(axis=0)
[ 1.98923924 2.43743744 22.90102603]
The run() method splits the computation over 10 processors and collates the results. When the parameter numberOfSamples is used, the regions selected for the computation of the coverage are not random. Instead, the genome is split into ‘number-of-samples’ equal parts and the start of each part is queried for its coverage. You can also compute coverage over selected regions by inputting a BED file.
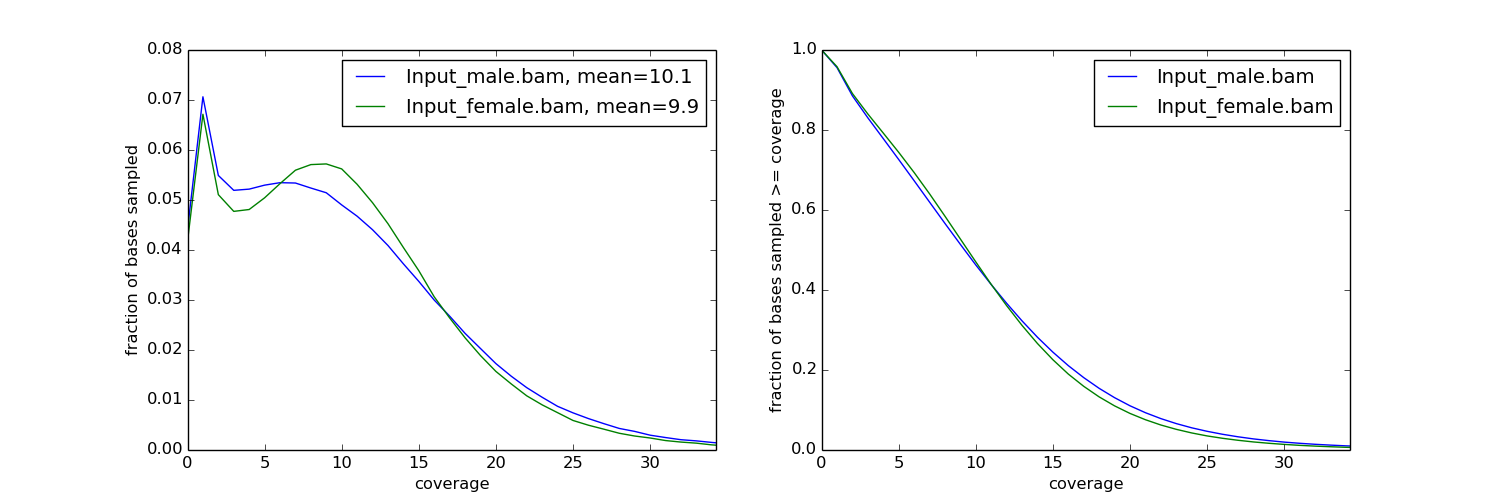
Now it is possible to make some diagnostic plots from the results:
fig, axs = plt.subplots(1, 2, figsize=(15,5))
# plot coverage
for col in res.T:
axs[0].plot(np.bincount(col.astype(int)).astype(float)/total_sites)
csum = np.bincount(col.astype(int))[::-1].cumsum()
axs[1].plot(csum.astype(float)[::-1] / csum.max())
axs[0].set_xlabel('coverage')
axs[0].set_ylabel('fraction of bases sampled')
# plot cumulative coverage
axs[1].set_xlabel('coverage')
axs[1].set_ylabel('fraction of bases sampled >= coverage')
Computing the FRiP score
The FRiP score is defined as the fraction of reads that fall into a peak and is often used as a measure of ChIP-seq quality. For this example, we need a BED file containing the peak regions. Such files are usually computed using a peak caller. Also, two bam files are going to be used, corresponding to two biological replicates.
bed_files = ["peaks.bed"]
cr = countReadsPerBin.CountReadsPerBin([bam_file1, bam_file2],
bedFile=bed_files,
numberOfProcessors=10)
reads_at_peaks = cr.run()
print reads_at_peaks
array([[ 322., 248.],
[ 231., 182.],
[ 112., 422.],
...,
[ 120., 76.],
[ 235., 341.],
[ 246., 265.]])
The result is a numpy array with a row for each peak region and a column for each BAM file.
reads_at_peaks.shape
(6295, 2)
Now, the total number of reads per peaks per bam file is computed:
total = reads_at_peaks.sum(axis=0)
Next, we need to find the total number of mapped reads in each of the bam files. For this we use the pysam module.
import pysam
bam1 = pysam.AlignmentFile(bam_file1)
bam2 = pysam.AlignmentFile(bam_file2)
Now, bam1.mapped and bam2.mapped contain the total number of mapped reads in each of the bam files, respectively.
Finally, we can compute the FRiP score:
frip1 = float(total[0]) / bam1.mapped
frip2 = float(total[1]) / bam2.mapped
print frip1, frip2
0.170030741997, 0.216740390353
Using mapReduce to sample paired-end fragment lengths
deepTools internally uses a map-reduce strategy, in which a computation is split into smaller parts that are sent to different processors. The output from the different processors is subsequently collated. The following example is based on the code available for bamPEFragmentSize.py
Here, we retrieve the reads from a BAM file and collect the fragment length. Reads are retrieved using pysam, and the read object returned contains the template_length attribute, which is the number of bases from the leftmost to the rightmost mapped base in the read pair.
First, we will create a function that can collect fragment lengths over a genomic position from a BAM file. As we will later call this function using mapReduce, the function accepts only one argument, namely a tuple with the parameters: chromosome name, start position, end position, and BAM file name.
import pysam
import numpy as np
def get_fragment_length(args):
chrom, start, end, bam_file_name = args
bam = pysam.AlignmentFile(bam_file_name)
f_lens_list = []
for fetch_start in range(start, end, 1000000):
# simply get the reads over a region of 10000 bases
fetch_end = min(end, fetch_start + 10000)
f_lens_list.append(np.array([abs(read.template_length)
for read in bam.fetch(chrom, fetch_start, fetch_end)
if read.is_proper_pair and read.is_read1]))
# concatenate all results
return np.concatenate(f_lens_list)
Now, we can use mapReduce to call this function and compute fragment lengths over the whole genome. mapReduce needs to know the chromosome sizes, which can be easily retrieved from the BAM file. Furthermore, it needs to know the size of the region(s) sent to each processor. For this example, a region of 10 million bases is sent to each processor using the genomeChunkLength parameter. In other words, each processor executes the same get_fragment_length function to collect data over different 10 million base regions. The arguments to mapReduce are the list of arguments sent to the function, besides the first obligatory three (chrom start, end). In this case only one extra argument is passed to the function, the BAM file name. The next two positional arguments are the name of the function to call (get_fragment_length) and the chromosome sizes.
import deeptools.mapReduce
bam = pysam.AlignmentFile(bamFile)
chroms_sizes = list(zip(bam.references, bam.lengths))
result = mapReduce.mapReduce([bam_file_name],
get_fragment_length,
chroms_sizes,
genomeChunkLength=10000000,
numberOfProcessors=20,
verbose=True)
fragment_lengths = np.concatenate(result)
print("mean fragment length {}".format(fragment_lengths.mean()))
print("median fragment length {}".format(np.median(fragment_lengths)))
0.170030741997, 0.216740390353