Glossary of NGS terms
Like most specialized fields, next-generation sequencing has inspired many an acronyms. We are trying to keep track of those Abbreviations that we heavily use. Do make us aware if something is unclear by opening an issue on github
Abbreviations
Reference genomes are usually referred to by their abbreviations, such as:
hg19 = human genome, version 19
mm9 = Mus musculus genome, version 9
dm3 = Drosophila melanogaster, version 3
ce10 = Caenorhabditis elegans, version 10
For a more comprehensive list of available reference genomes and their abbreviations, see the UCSC data base.
Acronym |
full phrase |
Synonyms/Explanation |
|---|---|---|
<ANYTHING>-seq |
-sequencing |
indicates that an experiment was completed by DNA sequencing using NGS |
ChIP-seq |
chromatin immunoprecipitation sequencing |
NGS technique for detecting transcription factor binding sites and histone modifications (see entry Input for more information) |
DNase |
deoxyribonuclease I |
DNase I digestion is used to determine active (“open”) chromatin regions |
HTS |
high-throughput sequencing |
next-generation sequencing, massive parallel short read sequencing, deep sequencing |
MNase |
micrococcal nuclease |
MNase digestion is used to determine sites with nucleosomes |
NGS |
next-generation sequencing |
high-throughput (DNA) sequencing, massive parallel short read sequencing, deep sequencing |
RPGC |
reads per genomic content |
normalize reads to 1x sequencing depth, sequencing depth is defined as: (mapped reads x fragment length) / effective genome size |
RPKM |
reads per kilobase per million reads |
normalize read numbers: RPKM (per bin) = reads per bin / ( mapped reads (in millions) x bin length (kb)) |
For a review of popular *-seq applications, see Zentner and Henikoff.
NGS and generic terminology
The following are terms that may be new to some:
bin
synonyms: window, region
A ‘bin’ is a subset of a larger grouping. Many calculations calculation are performed by first dividing the genome into small regions (bins), on which the calculations are actually performed.
Input
Control experiment typically done for ChIP-seq experiments
While ChIP-seq relies on antibodies to enrich for DNA fragments bound to a certain protein, the input sample should be processed exactly the same way, excluding the antibody. This allows one to account for biases introduced by sample handling and the general chromatin structure of the cells
read
synonym: tag
This term refers to the piece of DNA that is sequenced (“read”) by the sequencers. We try to differentiate between “read” and “DNA fragment” as the fragments that are put into the sequencer tend to be in the range of 200-1000 bases, of which only the first 50 to 300 bases are typically sequenced. Most of the deepTools will not only take these reads into account, but also extend them to match the original DNA fragment size. (The original size will either be given by you or, if you used paired-end sequencing, be calculated from the distance between the two read mates).
File Formats
Data obtained from next-generation sequencing data must be processed several times. Most of the processing steps are aimed at extracting only that information needed for a specific down-stream analysis, with redundant entries often discarded. Therefore, specific data formats are often associated with different steps of a data processing pipeline.
Here, we just want to give very brief key descriptions of the file, for elaborate information we will link to external websites. Be aware, that the file name sorting here is alphabetical, not according to their usage within an analysis pipeline that is depicted here:
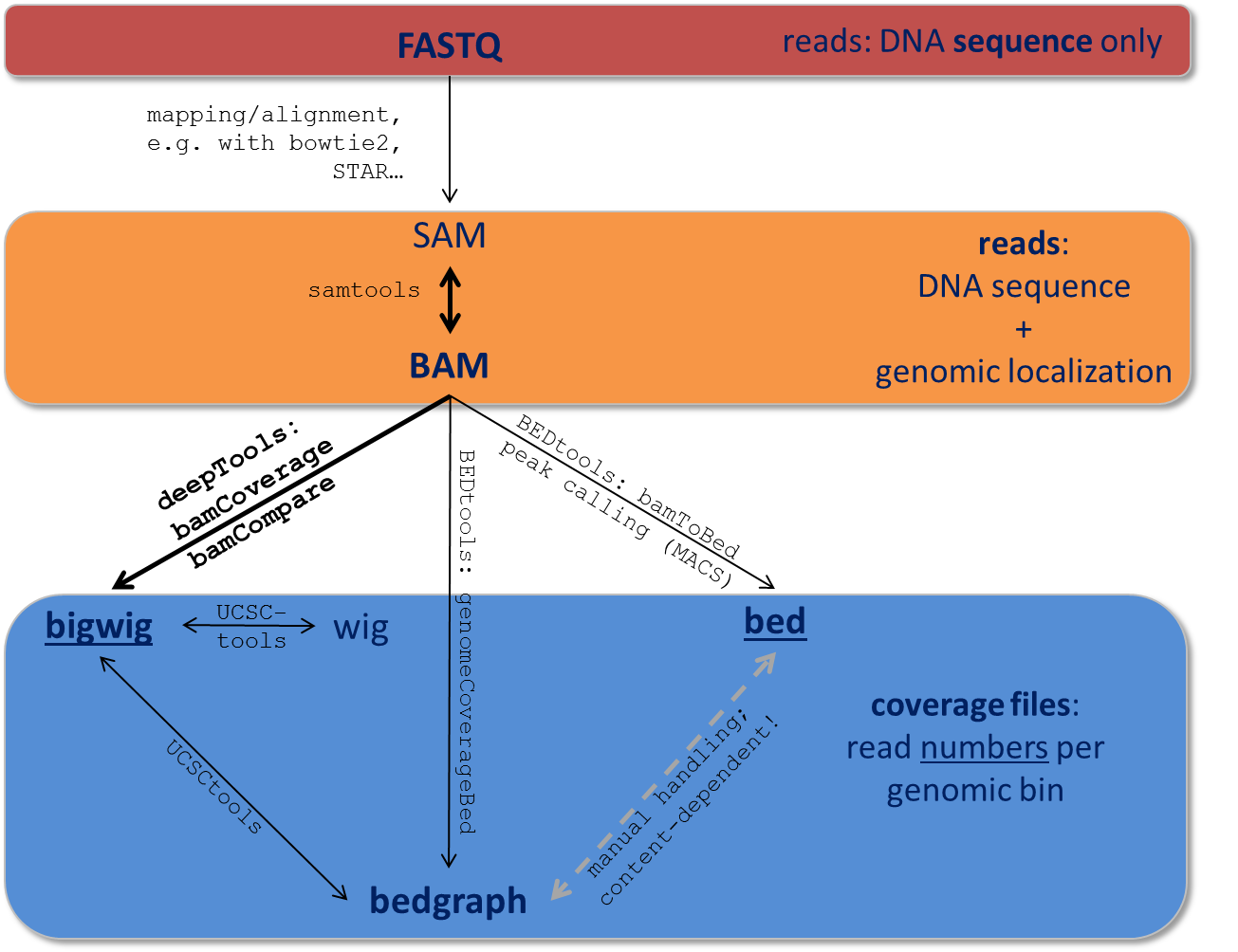
Follow the links for more information on the different tool collections mentioned in the figure:
samtools | UCSCtools | BEDtools |
2bit
BAM
typical file extension:
.bambinary file format (complement to SAM)
contains information about sequenced reads (typically) after alignment to a reference genome
- each line = 1 mapped read, with information about:
its mapping quality (how likelihood that the reported alignment is correct)
its sequencing quality (the probability that each base is correct)
its sequence
its location in the genome
etc.
highly recommended format for storing data
to make a BAM file human-readable, one can, for example, use the program samtools view
for more information, see below for the definition of SAM files
BED
typical file extension:
.bedtext file
used for genomic intervals, e.g. genes, peak regions etc.
the format can be found at UCSC
for deepTools, the first 3 columns are important: chromosome, start position of the region, end position of the genome
do not confuse it with the bedGraph format (although they are related)
example lines from a BED file of mouse genes (note that the start position is 0-based, the end-position 1-based, following UCSC conventions for BED files):
chr1 3204562 3661579 NM_001011874 Xkr4 - chr1 4481008 4486494 NM_011441 Sox17 - chr1 4763278 4775807 NM_001177658 Mrpl15 - chr1 4797973 4836816 NM_008866 Lypla1 +
bedGraph
typical file extension:
.bg,.bedGraphtext file
similar to BED file (not the same!), it can only contain 4 columns and the 4th column must be a score
again, read the UCSC description for more details
4 example lines from a bedGraph file (like BED files following the UCSC convention, the start position is 0-based, the end-position 1-based in bedGraph files):
chr1 10 20 1.5
chr1 20 30 1.7
chr1 30 40 2.0
chr1 40 50 1.8
bigWig
typical file extension:
.bw,.bigwigbinary version of a bedGraph or
wigfilecontains coordinates for an interval and an associated score
the score can be anything, e.g. an average read coverage
UCSC description for more details
FASTA
typical file extension:
.fastatext file, often gzipped (
.fasta.gz)very simple format for DNA/RNA or protein sequences, this can be anything from small pieces of DNA or proteins to an entire genome (most likely, you will get the genome sequence of your organism of interest in fasta format)
see the 2bit file format entry for a compressed alternative
example from wikipedia showing exactly one sequence:
>gi|5524211|gb|AAD44166.1| cytochrome b [Elephas maximus maximus]
LCLYTHIGRNIYYGSYLYSETWNTGIMLLLITMATAFMGYVLPWGQMSFWGATVITNLFSAIPYIGTNLV
EWIWGGFSVDKATLNRFFAFHFILPFTMVALAGVHLTFLHETGSNNPLGLTSDSDKIPFHPYYTIKDFLG
LLILILLLLLLALLSPDMLGDPDNHMPADPLNTPLHIKPEWYFLFAYAILRSVPNKLGGVLALFLSIVIL
GLMPFLHTSKHRSMMLRPLSQALFWTLTMDLLTLTWIGSQPVEYPYTIIGQMASILYFSIILAFLPIAGX
IENY
FASTQ
typical file extension:
.fastq,.fqtext file, often gzipped (–>
.fastq.gz)- contains raw read information – 4 lines per read:
read ID
base calls
additional information or empty line
sequencing quality measures - 1 per base call
note that there is no information about where in the genome the read originated from
example from the wikipedia page, which contains further information:
@read001 # read ID GATTTGGGGTTCAAAGCAGTATCGATCAAATAGTAAATCCATTTGTTCAACTCACAGTTT # read sequence + # usually empty line !''*((((***+))%%%++)(%%%%).1***-+*''))**55CCF>>>>>>CCCCCCC65 # ASCII-encoded quality scores
if you need to find out what type of ASCII-encoding your .fastq file contains, you can simply run FastQC – its summery file will tell you
SAM
typical file extension:
.samusually the result of an alignment of sequenced reads to a reference genome
contains a short header section (entries are marked by @ signs) and an alignment section where each line corresponds to a single read (thus, there can be millions of these lines)
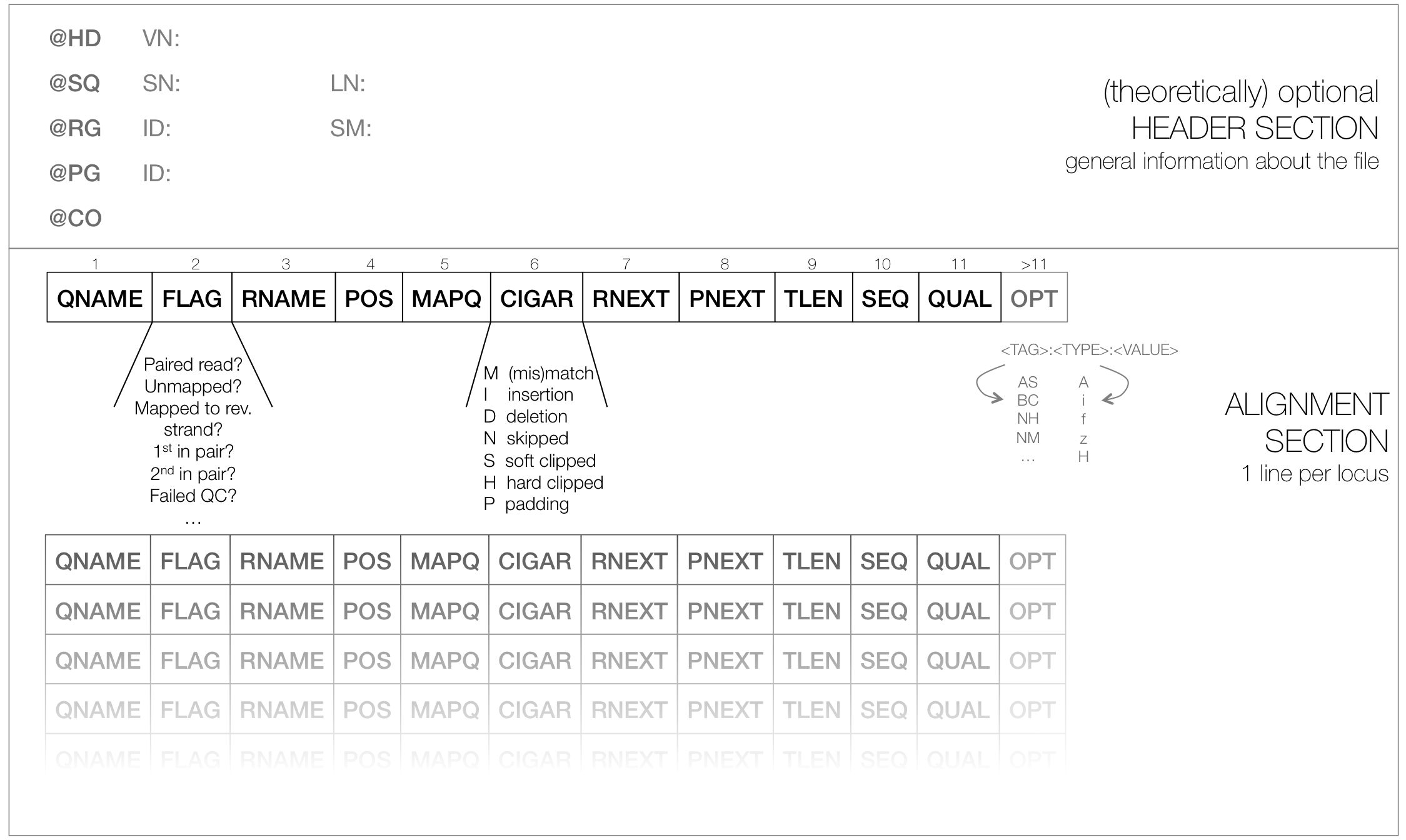
SAM header section
tab-delimited lines, beginning with @, followed by tag:value pairs
tag = two-letter string that defines the content and the format of value
SAM alignment section
each line contains information about its mapping quality, its sequence, its location in the genome etc.
r001 163 chr1 7 30 8M2I4M1D3M = 37 39 TTAGATAAAGGATACTG * r002 0 chr1 9 30 3S6M1P1I4M * 0 0 AAAAGATAAGGATA *the flag in the second field contains the answer to several yes/no assessments that are encoded in a single number
for more details on the flag, see this thorough explanation or this more technical explanation
the CIGAR string in the 6th field represents the types of operations that were needed in order to align the read to the specific genome location:
insertion
deletion (small deletions denoted with D, bigger deletions, e.g., for spliced reads, denoted with N)
clipping (deletion at the ends of a read)
Warning
Although the SAM/BAM format is rather meticulously defined and documented, whether an alignment program will produce a SAM/BAM file that adheres to these principles is completely up to the programmer. The mapping score, CIGAR string, and particularly, all optional flags (fields >11) are often very differently defined depending on the program. If you plan on filtering your data based on any of these criteria, make sure you know exactly how these entries were calculated and set!