computeGCBias
Computes the GC-bias using Benjamini’s method [Benjamini & Speed (2012). Nucleic Acids Research, 40(10). doi: 10.1093/nar/gks001]. The GC-bias is visualized and the resulting table can be used tocorrect the bias with correctGCBias.
usage: computeGCBias -b file.bam --effectiveGenomeSize 2150570000 -g mm9.2bit -l 200 --GCbiasFrequenciesFile freq.txt
help: computeGCBias -h / computeGCBias --help
Required arguments
- --bamfile, -b
Sorted BAM file.
- --effectiveGenomeSize
The effective genome size is the portion of the genome that is mappable. Large fractions of the genome are stretches of NNNN that should be discarded. Also, if repetitive regions were not included in the mapping of reads, the effective genome size needs to be adjusted accordingly. A table of values is available here: http://deeptools.readthedocs.io/en/latest/content/feature/effectiveGenomeSize.html .
- --genome, -g
Genome in two bit format. Most genomes can be found here: http://hgdownload.cse.ucsc.edu/gbdb/ Search for the .2bit ending. Otherwise, fasta files can be converted to 2bit using the UCSC programm called faToTwoBit available for different plattforms at http://hgdownload.cse.ucsc.edu/admin/exe/
- --GCbiasFrequenciesFile, -freq, -o
Path to save the file containing the observed and expected read frequencies per %GC-content. This file is needed to run the correctGCBias tool. This is a text file.
Optional arguments
- --fragmentLength, -l
Fragment length used for the sequencing. If paired-end reads are used, the fragment length is computed based from the bam file
- --sampleSize
Number of sampling points to be considered. (Default: 50000000.0)
- --extraSampling
BED file containing genomic regions for which extra sampling is required because they are underrepresented in the genome.
- --version
show program’s version number and exit
- --region, -r
Region of the genome to limit the operation to - this is useful when testing parameters to reduce the computing time. The format is chr:start:end, for example –region chr10 or –region chr10:456700:891000.
- --blackListFileName, -bl
A BED or GTF file containing regions that should be excluded from all analyses. Currently this works by rejecting genomic chunks that happen to overlap an entry. Consequently, for BAM files, if a read partially overlaps a blacklisted region or a fragment spans over it, then the read/fragment might still be considered. Please note that you should adjust the effective genome size, if relevant.
- --numberOfProcessors, -p
Number of processors to use. Type “max/2” to use half the maximum number of processors or “max” to use all available processors. (Default: 1)
- --verbose, -v
Set to see processing messages.
Diagnostic plot options
- --biasPlot
If given, a diagnostic image summarizing the GC-bias will be saved.
- --plotFileFormat
Possible choices: png, pdf, svg, eps, plotly
image format type. If given, this option overrides the image format based on the plotFile ending. The available options are: “png”, “eps”, “pdf”, “plotly” and “svg”
- --regionSize
To plot the reads per %GC over a regionthe size of the region is required. By default, the bin size is set to 300 bases, which is close to the standard fragment size for Illumina machines. However, if the depth of sequencing is low, a larger bin size will be required, otherwise many bins will not overlap with any read (Default: 300)
Background
computeGCBias is based on a paper by Benjamini and Speed.
The basic assumption of the GC bias diagnosis is that an ideal sample should show a uniform distribution of sequenced reads across the genome, i.e. all regions of the genome should have similar numbers of reads, regardless of their base-pair composition.
In reality, the DNA polymerases used for PCR-based amplifications during the library preparation of the sequencing protocols prefer GC-rich regions. This will influence the outcome of the sequencing as there will be more reads for GC-rich regions just because of the DNA polymerase’s preference.
computeGCbias will first calculate the expected GC profile by counting the number of DNA fragments of a fixed size per GC fraction where GC fraction is defined as the number of G’s or C’s in a genome region of a given length.
The result is basically a histogram depicting the frequency of DNA fragments for each type of genome region with a GC fraction between 0 to 100 percent. This will be different for each reference genome, but is independent of the actual sequencing experiment.
The profile of the expected DNA fragment distribution is then compared to the observed GC profile, which is generated by counting the number of sequenced reads per GC fraction.
In an ideal experiment, the observed GC profile would, of course, look like the expected profile.
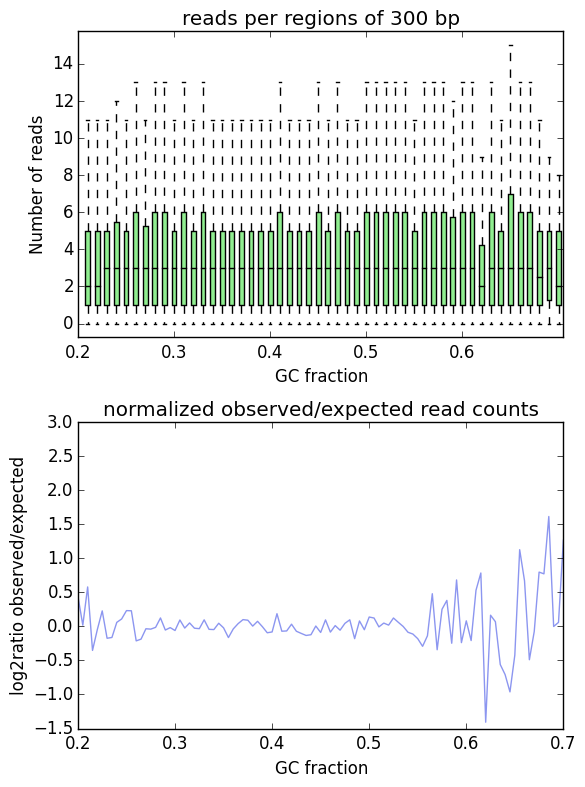
This is indeed the case when applying computeGCBias to simulated reads.
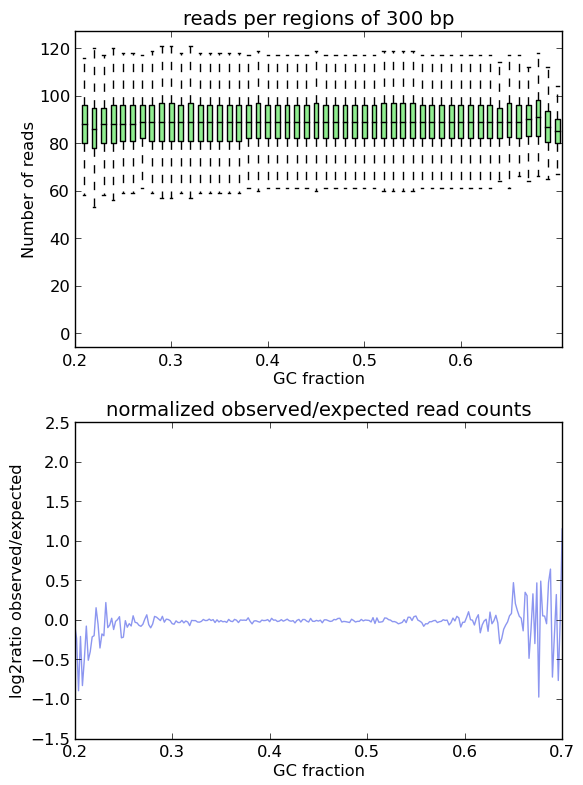
As you can see, both plots based on simulated reads do not show enrichments or depletions for specific GC content bins, there is an almost flat line around the log2ratio of 0 (= ratio(observed/expected) of 1). The fluctuations on the ends of the x axis are due to the fact that only very, very few regions in the Drosophila genome have such extreme GC fractions so that the number of fragments that are picked up in the random sampling can vary.
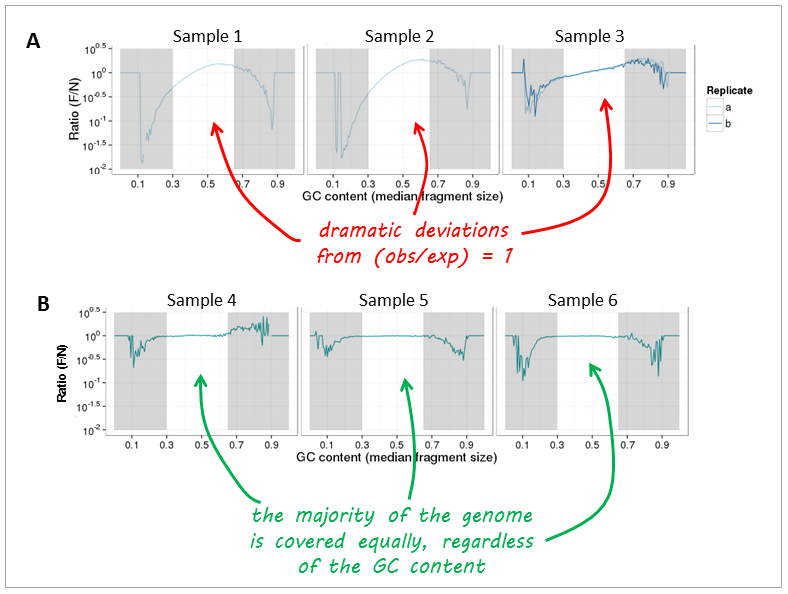
Now, let’s have a look at real-life data from genomic DNA sequencing. Panels A and B can be clearly distinguished and the major change that took place between the experiments underlying the plots was that the samples in panel A were prepared with too many PCR cycles and a standard polymerase whereas the samples of panel B were subjected to very few rounds of amplification using a high fidelity DNA polymerase.
Note
The expected GC profile depends on the reference genome as different organisms have very different GC contents. For example, one would expect more fragments with GC fractions between 30% to 60% in mouse samples (average GC content of the mouse genome: 45 %) than for genome fragments from, for example, Plasmodium falciparum (average genome GC content P. falciparum: 20%).
Excluding regions from the read distribution calculation
In some cases, it will make sense to exclude certain regions from the calculation of the read distributions to increase the accuracy of the computation. There are several kinds of regions that are either not expected to show a read distribution matching the background or where the uncertainty of the reference genome might be too big. Please consider the following points:
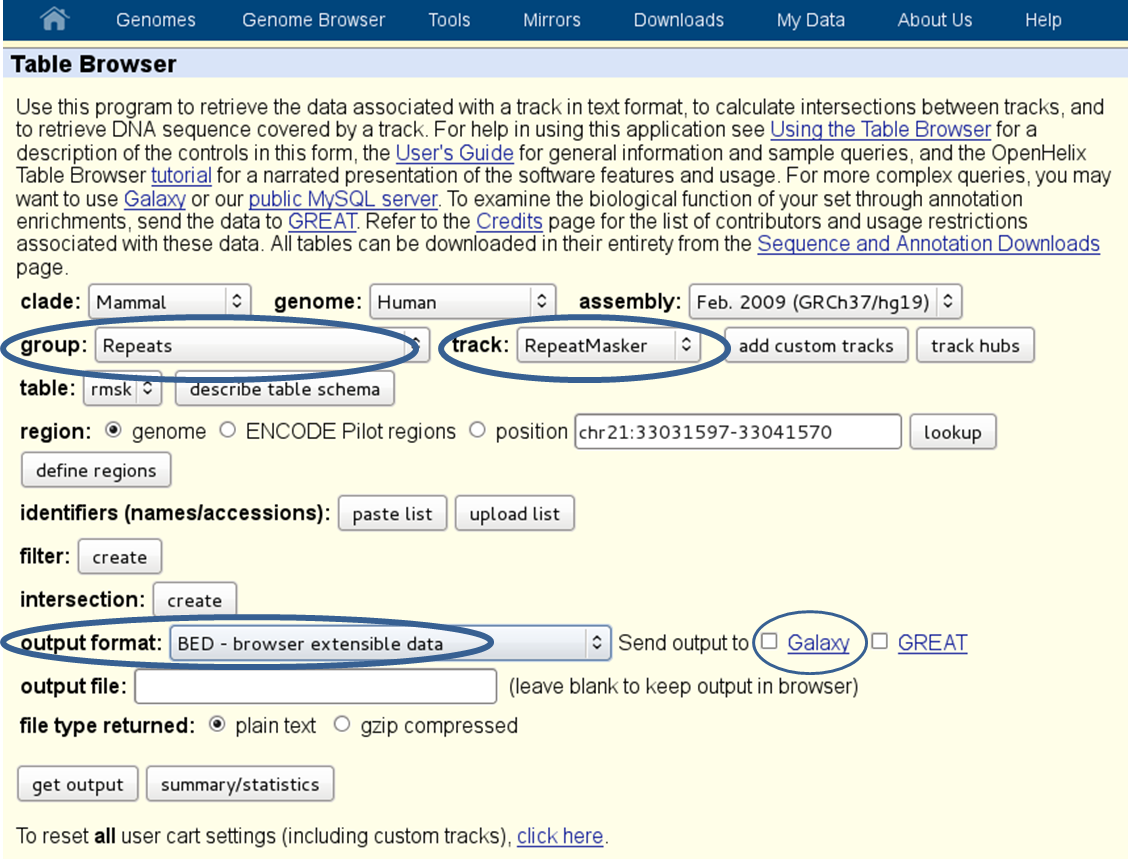
repetitive regions: if multi-reads (reads that map to more than one genomic position) were excluded from the [BAM][] file, it will help to exclude known repetitive regions. You can get BED files of known repetitive regions from UCSC Table Browser (see the screenshot below for an example of human repetitive elements).
regions of low mappability: these are regions where the mapping of the reads notoriously fails and we recommend to exclude known regions with mappability issues from the GC computation. You can download the mappability tracks for different read lengths from UCSC, e.g. for mouse and human. In the github deepTools folder “scripts”, you can find a shell script called
mappabilityBigWig_to_unmappableBed.shwhich will turn the [bigWig][] mappability file from UCSC into a BED file.ChIP-seq peaks: in ChIP-seq samples it is expected that certain regions should show more reads than expected based on the background distribution, therefore it makes absolute sense to exclude those regions from the GC bias calculation. We recommend to run a simple, non-conservative peak calling on the uncorrected [BAM][] file first to obtain a BED file of peak regions that should then subsequently be supplied to
computeGCbias.
Usage example
$ computeGCBias -b H3K27Me3.bam --effectiveGenomeSize 2695000000
--genome genome.2bit -l 200 -freq freq_test.txt
--region X --biasPlot test_gc.png